
La hiperglicinemia no cetósica (HNC) es una enfermedad autosómica recesiva por una deficiencia en la división del sistema enzimático de la glicina mitocondrial (GCS) que presenta un complejo de cuatro proteínas codificadas en cuatro cromosomas diferentes. Defectos genéticos en cualquiera de las tres primeras han sido descritos en pacientes con diferentes fenotipos de HNC1–3. Hasta un 80% de pacientes tienen mutaciones diferentes en el gen glicina-decarboxilasa (GLDC)4 con supresión del gen GLDC en el 20%5,6. El exceso de glicina ocasiona una sobreestimulación de los receptores inhibidores de glicina, siendo responsable de la aparición de hipotonía y apnea, y sobre los receptores excitatorios del N-metil-D-aspartato (NMDAR) provocando un daño neuronal citotóxico por estimulación de receptores adicionales glicinérgicos3.
La clínica y una relación de glicina en líquido cefalorraquídeo (LCR)/plasma>0,08 y la ausencia de la aciduria orgánica son esenciales para el diagnóstico. Su confirmación requiere la medición de la actividad del enzima GCS en el tejido hepático, o el estudio genético en fibroblastos6. El tratamiento pretende la reducción de los niveles plasmáticos de glicina por la conjugación de benzoato de sodio y por la administración de antagonistas del receptor de NMDA como la ketamina o el dextrometorfano3,7. Sin embargo, las consecuencias neurológicas de la HNC son devastadoras, incluso si el tratamiento se instaura precozmente3,8.
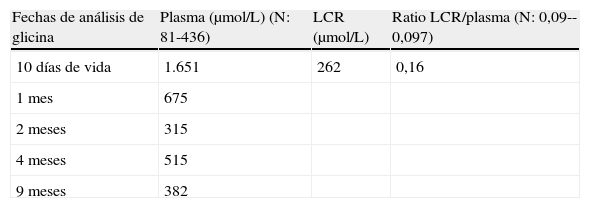
Se describe un caso clínico de un neonato con una forma clásica de HNC que presenta una nueva mutación: Recién nacido a término que ingresó por hipotonía severa presentando a los 2 días movimientos mioclónicos y apnea recurrente evolucionando al coma. Se demostró agenesia del cuerpo calloso con atrofia cerebral severa y un patrón brote-supresión en el electroencefalograma. Ante estos hallazgos se sospechó HNC, demostrándose altos niveles de glicina en plasma y LCR, y una alta proporción de glicina LCR/plasma (tabla 1). El análisis genético en fibroblastos demostró una mutación en homocigosis del gen GLDC con una única sustitución en el nucleótido 1742 del exón 15, dando lugar a un cambio de prolina por arginina en la posición 581: (c.1742C> G/p.P581R). Aunque no se demostró consanguinidad en los progenitores, ambos padres presentaron la misma mutación en heterocigosis. El tratamiento se inició con fármacos tradicionales como benzoato de sodio y dextrometorfano a los 11 días de vida, desapareciendo los movimientos mioclónicos y el estado de coma. Se asoció una dieta restringida en proteínas, glicina y otros aminoácidos convertibles en esta como la treonina y la serina con ganancia ponderal adecuada. Se mantuvo una situación neurológica severa, pero sin convulsiones y con niveles plasmáticos de glicina dentro de rangos normales. No se registraron más episodios de apnea, y progresivamente adquirió llanto y deglución. A los 9 meses, ingería la mayor parte de la alimentación por vía oral con mioclonias pero sin crisis convulsivas, y atrofia cortical progresiva con retraso psicomotor grave.
Niveles plasmáticos y en líquido cefalorraquídeo de glicina para el diagnóstico y la evolución de un paciente pediátrico con hiperglicinemia no cetósica
| Fechas de análisis de glicina | Plasma (μmol/L) (N: 81-436) | LCR (μmol/L) | Ratio LCR/plasma (N: 0,09--0,097) |
| 10 días de vida | 1.651 | 262 | 0,16 |
| 1 mes | 675 | ||
| 2 meses | 315 | ||
| 4 meses | 515 | ||
| 9 meses | 382 |
LCR: líquido cefalorraquídeo.
Es importante sospechar este error innato del metabolismo en recién nacidos con episodios de apnea, hipotonía y evolución a coma3, e iniciar el estudio precozmente, ya que la HNC puede no detectarse en el screening realizado mediante tándem masas9. Se han descrito síntomas clínicos muy heterogéneos en esta enfermedad, algunos de ellos por mutaciones del gen GLDC identificados en estado heterocigoto, homocigotos o compuestos4. Algunas de las mutaciones están asociadas con una actividad residual de la glicina decarboxilasa y la identificación de estas y la asociación a formas atípicas puede ayudar a predecir qué pacientes pueden tener un fenotipo más leve, así como aquellos que puedan responder a la intervención terapéutica temprana. No obstante, hay que considerar que la glicina posiblemente también induce una lesión prenatal grave en el sistema nervioso central3,5.
Esta nueva mutación ha podido condicionar la evolución referida en este niño. No se ha descrito anteriormente una variante polimórfica en la unión de la glicina. Por otra parte, esta mutación en homocigosis afecta a una posición altamente conservada en la evolución filogenética y tiene una alta probabilidad de ser patógena y de ser la responsable de la enfermedad del paciente. De este estudio se puede deducir que esta nueva mutación en GLDC en homocigosis da lugar a una forma neonatal de HNC en la que las crisis convulsivas han sido muy infrecuentes y con una inicial respuesta al tratamiento instaurado. No obstante, debemos ser cautos al describir fenotipos asociados a nuevas mutaciones que se refieran a un único caso. El conocer la etiología genética y poder aconsejar a las familias es fundamental para prevenir nuevos casos y tener en cuenta consideraciones bioéticas en el tratamiento de esta enfermedad10.
