


Bajo el término de raquitismo hipofosfatémico (RH) se engloban un conjunto de síndromes genéticos caracterizados por: afectación de la mineralización esquelética, hiperfosfaturia e hipofosfatemia, en ausencia de deficiencia de vitamina D1,2. Este cuadro clínico es causado por un incremento de los niveles del factor de crecimiento fibroblástico (FGF)-23 quien, actuando sobre su co-receptor alfa-Klotho, impide una reabsorción adecuada del fosfato en el túbulo renal proximal e interfiere en la hidroxilación renal de la vitamina D1–3. Por este motivo, el tratamiento actual del RH consiste en la suplementación de fosfato y de formas bioactivas de vitamina D4.
Los genes implicados y el modelo de herencia de estos síndrome pueden ser diversos, siendo el mejor caracterizado el RH dominante ligado al cromosoma X, resultante de mutaciones inactivantes en el gen phosphate regulating endopeptidase analog, X-linked (PHEX), que codifica para una metaloproteasa de membrana moduladora de la expresión y degradación del FGF-235. Aún así, incluso dentro de este subgrupo, existe una notable variabilidad clínica y evolutiva, con respuesta heterogénea al tratamiento, como ilustramos con los siguientes casos:
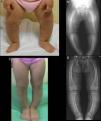
Caso 1Niña de 21 meses nacida a término, con antropometría adecuada, de padres sanos, no consanguíneos, sin antecedentes familiares de afección esquelética.
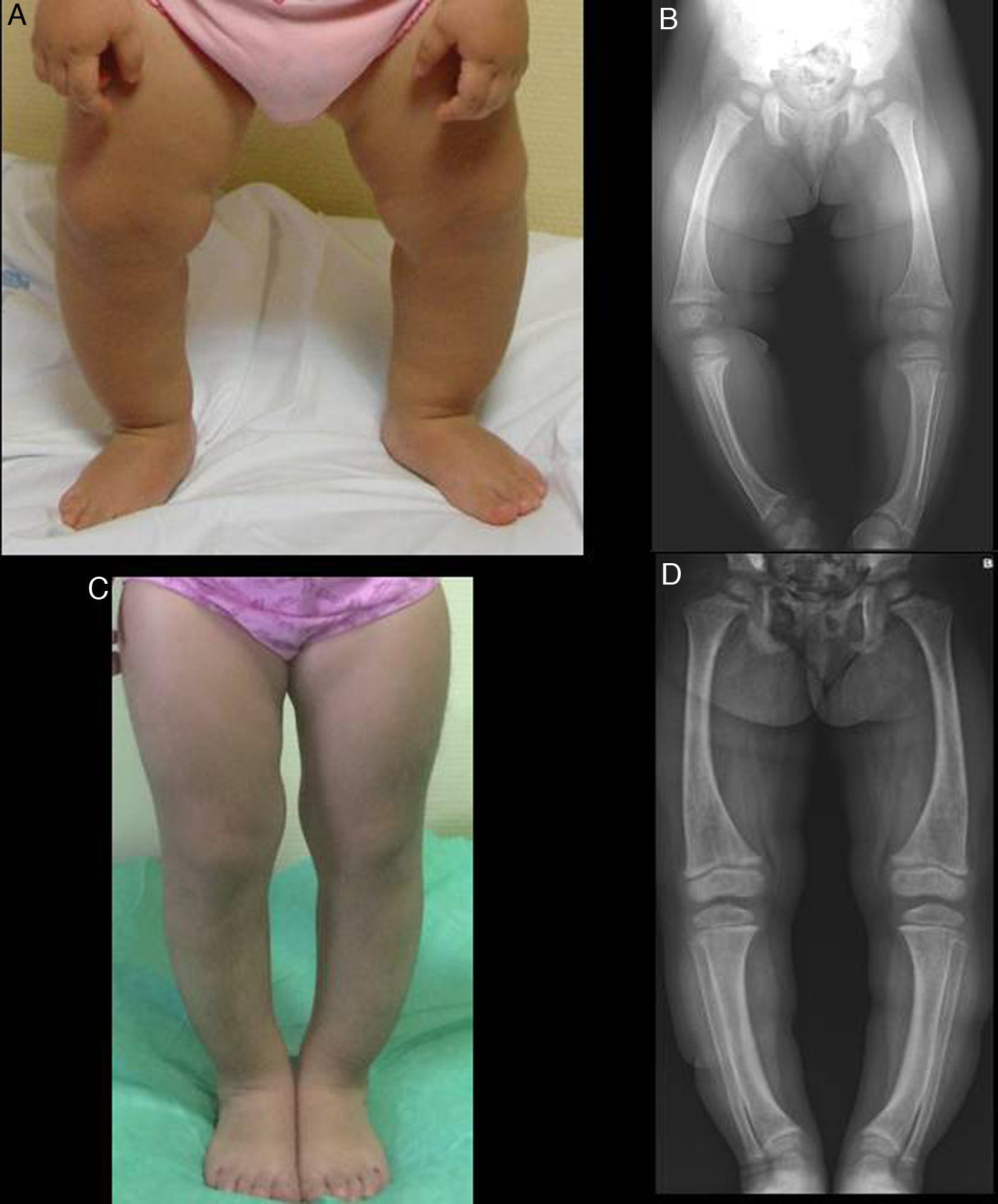
En la exploración presenta genu varum simétrico (fig. 1A), que determina una marcha «anadeante», confirmándose radiológicamente por la presencia de: tibias varas, desflecamiento de la línea metafisaria distal de ambos fémures y tibias e imágenes «en copa» evidentes en ambos peronés (fig. 1B), así como en cúbito y radio (con leve ensanchamiento de la extremidad distal de antebrazos).
El perfil bioquímico sérico (en ausencia de deficiencia proteica) determinó la existencia de niveles normales de magnesio y calcio, tanto total, como iónico, junto con una hipofosfatemia grave (2,3mg/dl; 4,5-6,7) y una elevación de la actividad de la fosfatasa alcalina (528U/l; 122-290). El cálculo de la tasa de reabsorción tubular de fosfatos (RTP) en orina de una micción no resultó aclaratorio (83,2%), pero sí el realizado tras recogida de orina de 24h, que estaba claramente disminuido (67,1%).
Se observaron niveles normales de 25(OH)-vitamina D (27,1ng/dl) y elevados de 1,25 di (OH)-vitamina D (123pg/dl; 16-56) y PTH intacta (106pg/ml; 11-67). Ante la sospecha de un raquitismo hipofosfatémico, se determinaron los niveles séricos de FGF-23, que se encontraban por encima del límite de detección del ensayo (>426U/ml).
El diagnóstico etiológico se estableció tras el hallazgo, mediante secuenciación, de la mutación de novo c.2445 G>A (Trp627X), en heterocigosis, en el exón 22 de PHEX (previamente no descrita).
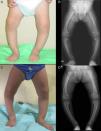
Caso 2Niña de 25 meses nacida a término, con antropometría adecuada, de padres sanos, no consanguíneos, sin antecedentes familiares de afección esquelética.
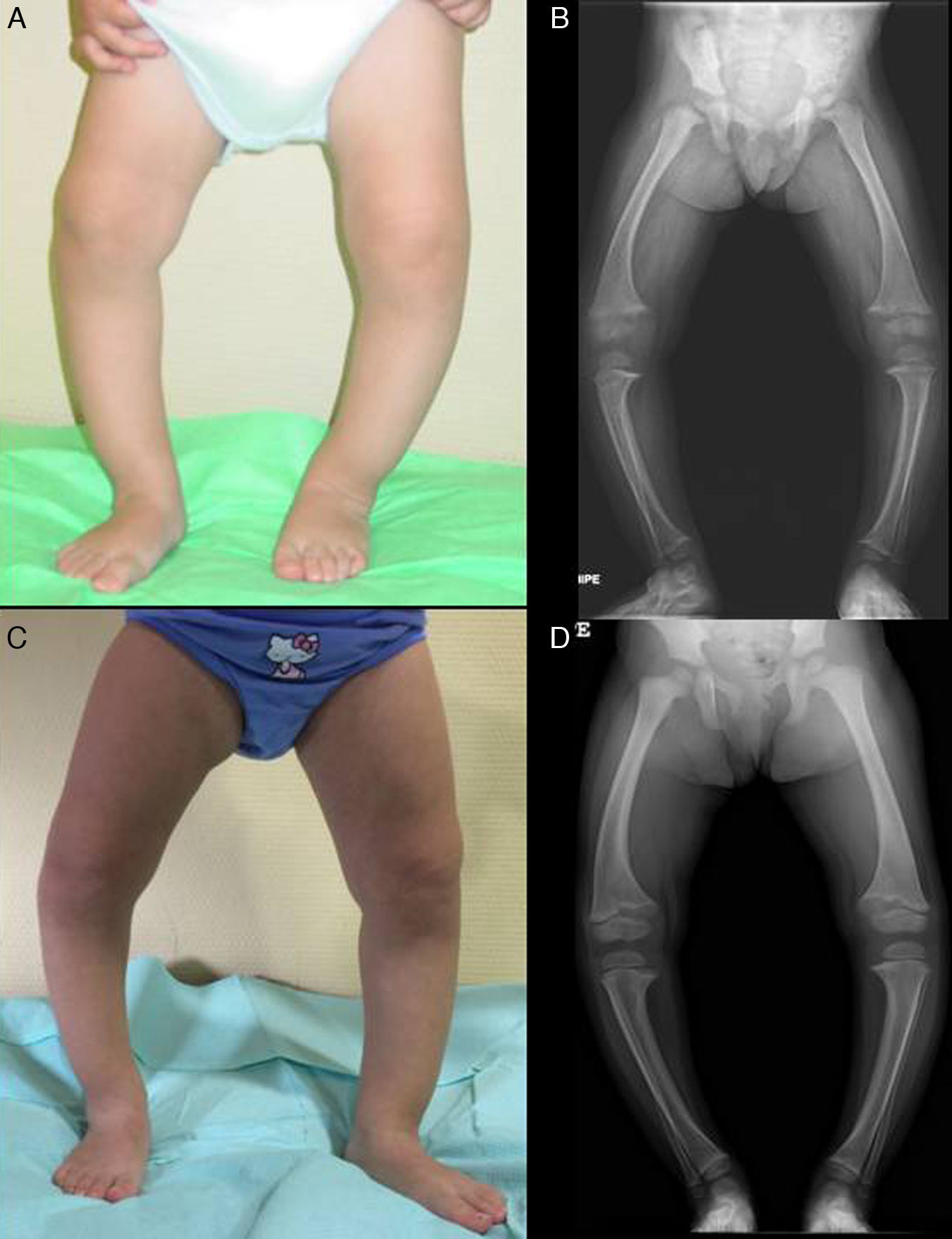
Presenta un grave genu varum asimétrico (predominio derecho) (fig. 2A), con marcha «anadeante» y con signos radiológicos similares a los descritos en el caso 1 (fig. 2B), y con mayor ensanchamiento de las extremidades distales de antebrazos.
Asimismo presentaba normocalcemia y normomagnesemia, con hipofosfatemia (2,9mg/dl; 4,5-6,7), hiperfosfatasemia (446U/l; 122-290) y RTP normal en orina de una micción (93,9%), y claramente disminuida en orina de 24h (67,5%).
También comparte con el caso 1 los niveles elevados de PTH intacta (116pg/ml; 11-67) y normales de 25(OH)-vitamina D (37,2ng/dl), si bien sus niveles de 1,25 di (OH)-vitamina D eran, asimismo, normales (55pg/dl; 16-56). La presencia de niveles séricos elevados de FGF-23, [253U/ml; VN<146) y el hallazgo de la mutación c.2301 G>A (Gly 579 Arg), en heterocigosis, en el exón 17 de PHEX, confirmaron el diagnóstico.
En ambos casos se pautó tratamiento con suplementos de fosfato oral y de formas bioactivas de vitamina D. Tras 2 años de tratamiento, en el caso 1 se observó una paulatina normalización de la fosforemia (3,6mg/dl) y de los niveles de PTH y 1,25 di (OH)-vitamina D, acompañados de una evidente mejoría clínica y radiológica (figs. 1C y 1D), si bien comenzó a desarrollar signos ecográficos sugerentes de nefrocalcinosis. En el caso 2, por el contrario, y pese a referir un adecuado cumplimiento terapéutico, se produjo un empeoramiento de la hipofosforemia (con valores repetidamente inferiores a 2,5mg/dl; mínimo: 2,3) y actividad sérica de fosfatasa alcalina persistentemente elevada (superior a 400U/l) pese al incremento paulatino de dosis de fosfato sódico hasta un máximo de 960mg de fósforo/día (81mg/kg). Asimismo, la evolución clínica y radiológica (figs. 2C y 2D) fueron insatisfactorias, requiriendo hemiepifisiodesis lateral externa en las metáfisis tibiales y femorales de ambas rodillas.
Esta respuesta dispar al tratamiento y la posibilidad de desarrollo de nefrocalcinosis hace atractiva la novedosa estrategia terapéutica, basada en el empleo de anticuerpos monoclonales frente al FGF-23, con resultados prometedores4 en pacientes adultos y ensayos activos (fase II y III) en pacientes pediátricos, tal y como se desarrolla en el artículo editorial que acompaña a esta carta científica6.
