


The term hypophosphatemic rickets (HR) encompasses a group of genetic diseases characterised by impairment of bone mineralisation, hyperphosphaturia and hypophosphatemia in the absence of vitamin D deficiency.1,2 These features are caused by increased levels of fibroblast growth factor 23 (FGF23), which, through interaction with its receptor α-klotho, prevents adequate reabsorption of phosphate in the proximal tubule and interferes with the hydroxylation of vitamin D in the kidney.1–3 Based on this, the current treatment for HR consists in supplementation with phosphate and bioactive forms of vitamin D.4
The inheritance patterns and genes involved in these syndromes vary. The syndrome that has been described most thoroughly to date is X-linked dominant HR, which results from inactivating mutations of PHEX (phosphate regulating endopeptidase homolog, X-linked), a gene encoding a cell-surface metalloprotease that regulates the expression and degradation of FGF23.5 Still, there is considerable variability in clinical manifestations and outcomes within this subgroup, and patients respond differently to treatment, which is illustrated by the cases presented here:
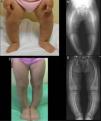
Case 1Girl aged 21 months born full term with normal anthropometric measurements to healthy, nonconsanguineous parents and with no family history of skeletal disease.
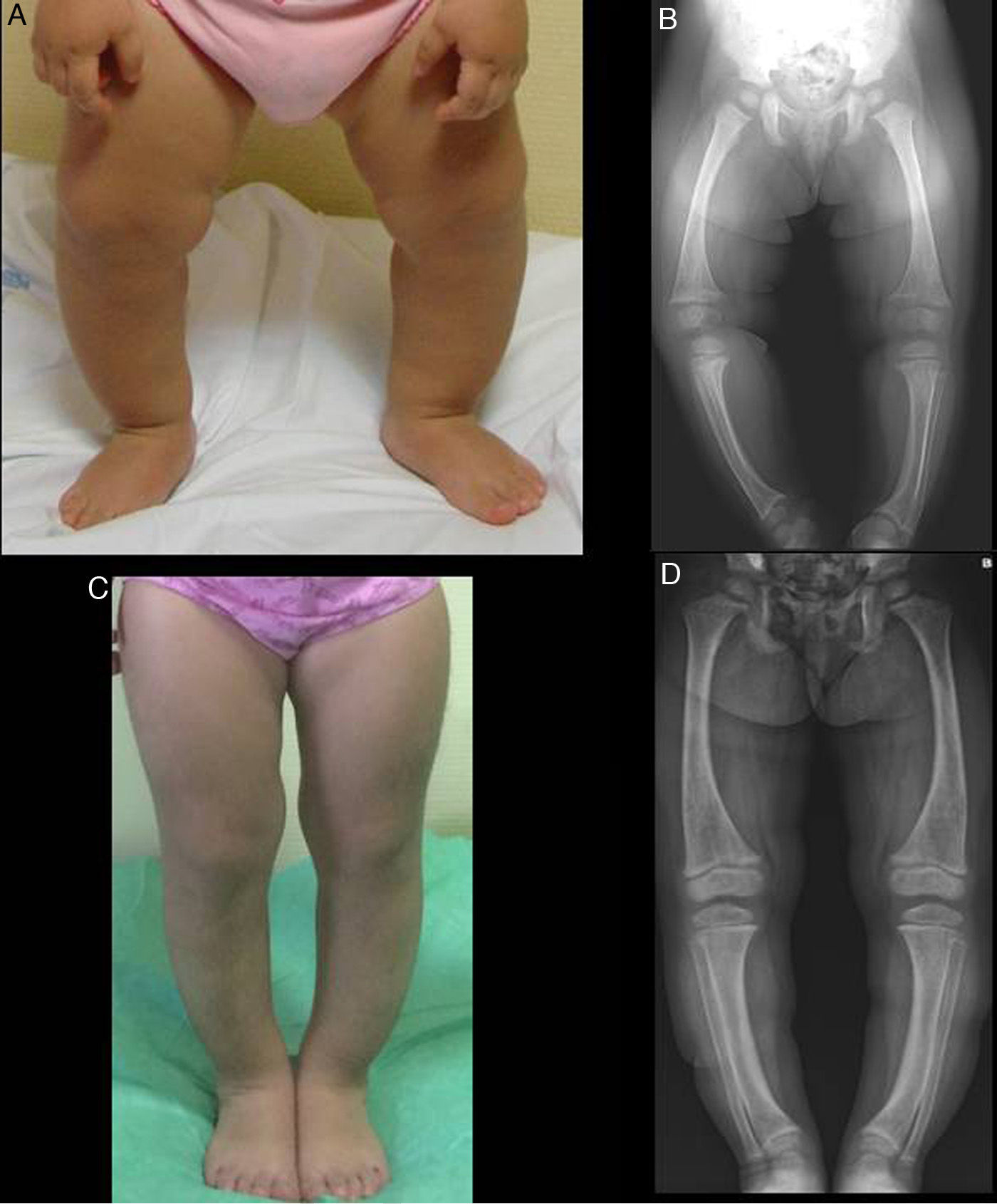
The physical examination found a symmetrical genu varum (Fig. 1A) associated with a waddling gait, confirmed by radiographic findings: varus of the tibias, fraying of the distal metaphyseal region of both femurs and tibias, and cupping of both fibulas (Fig. 1B), the ulnas and the radii (with a mild widening of the distal end of the forearms).
The blood chemistry panel (in the absence of protein deficiency) showed normal calcium and magnesium levels (ionised and total), along with severe hypophosphatemia (2.3mg/dL; normal range, 4.5–6.7) and increased alkaline phosphatase activity (528U/L; normal range, 122–290). The value of the rate of tubular phosphate reabsorption (TPR) obtained from a single-void urine sample was not indicative (83.2%), as opposed to the value obtained from the 24-h urine sample, which was clearly decreased (67.1%).
The panel showed normal levels of 25-hidroxyvitamin D (27.1ng/dL) and elevated levels of 1,25-dihidroxyvitamin D (123pg/dL; normal range, 16–56) and intact PTH (106pg/mL; normal range, 11–67). Hypophosphatemic rickets was suspected, leading to the determination of the serum FGF23 level, which exceeded the upper detection limit of the assay (>426U/mL).
The etiological diagnosis was made after gene sequencing found a de novo heterozygous mutation in exon 22 of PHEX (c.2445G>A [W627X]) that had not been previously described in the literature.
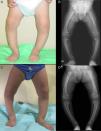
Case 2Girl aged 25 months born full term with normal anthropometric measurements to healthy nonconsanguineous parents and no family history of skeletal disease.
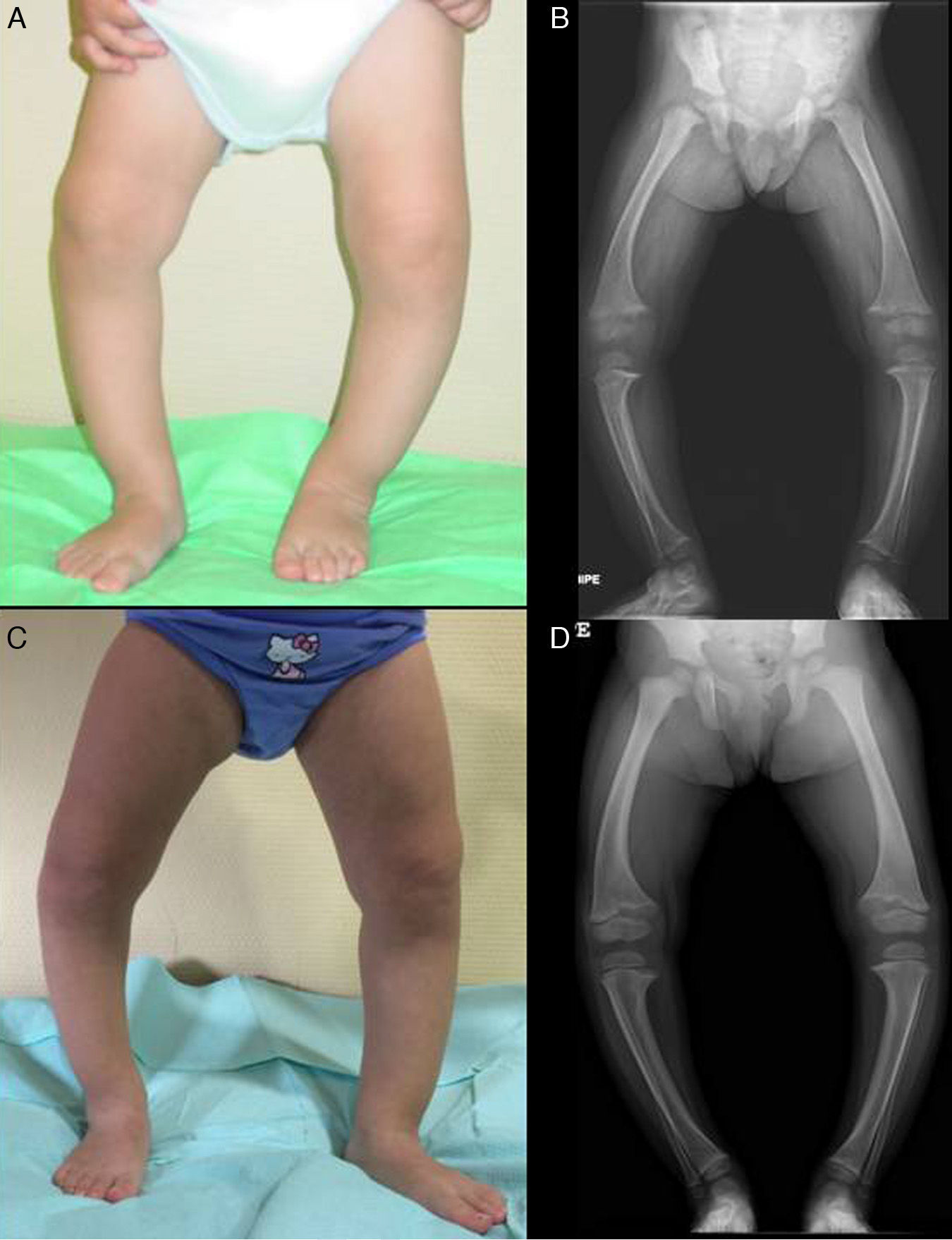
The patient presented with severe asymmetrical genu varum with the right side more affected than the left (Fig. 2A), a waddling gait and radiographic findings similar to those of case 1 (Fig. 2B), with a greater widening of the distal ends of the forearms.
The patient had normal calcium and magnesium levels, hypophosphatemia (2.9mg/dL, normal range, 4.5–6.7), hyperphosphatasemia (446U/L, normal range, 122–290), a normal rate of TPR in a single-void urine test (93.9%), and a clearly decreased TPR rate in the 24-h urine test (67.5%).
Also consistently with case 1, the patient had elevated levels of intact PTH (116pg/mL; normal range, 11–67) and normal levels of 25-hidroxyvitamin D (37.2ng/dL), although her levels of 1,25-dihidroxyvitamin D were also normal (55pg/dL; normal range, 16–56). The presence of an elevated serum FGF23 level (253U/mL; normal range <146) and the detection of a c.2301G>A (G579R) heterozygous mutation in exon 17 of PHEX confirmed the diagnosis.
Both cases were treated with oral phosphate and bioactive forms of vitamin D. After two years of treatment, case 1 had shown gradual normalisation of serum levels of phosphorus (3.6mg/dL), PTH and 1,25-dihidroxyvitamin D, along with evident clinical and radiographic improvement (Fig. 1C and D), although the patient started to show sonographic signs suggestive of nephrocalcinosis. Conversely, in case 2, despite the self-reported adherence to treatment, the hypophosphoremia worsened (with values repeatedly below 2.5mg/dL; lowest value, 2.3mg/dL) and persistently elevated alkaline phosphatase activity (greater than 400U/L) despite the gradual increase of the sodium phosphate dose to a maximum of 960mg of phosphorus a day (81mg/kg). Furthermore, the clinical and radiographic outcomes were poor (Fig. 2C and D), and the patient required lateral external hemyepiphysiodesis in the tibial and femoral metaphyses of both knees.
Given the different response to treatment and the possibility of developing nephrocalcinosis, the new therapeutic approach based on the use of anti-FGF23 monoclonal antibodies is very attractive, and it has showed promising results4 in adult patients and in ongoing trials (Phase II and III) in paediatric patients, as explained in the editorial that accompanies this scientific letter.6
Please cite this article as: Martos Moreno GÁ, Aparicio C, de Lucas C, Gil Peña H, Argente J. Raquitismo hipofosfatémico ligado a X por mutaciones en PHEX: variabilidad clínica y evolutiva. An Pediatr (Barc). 2016;85:41–43
