





La punta-onda continua durante el sueño lento (POCS) es un trazado electroencefalográfico característico, que aparece en la infancia y que en ocasiones condiciona un deterioro cognitivo. Este patrón electroencefalográfico puede aparecer tanto en determinados síndromes epilépticos como en la evolución de epilepsias idiopáticas y sintomáticas. El objetivo de nuestro estudio es analizar las características epidemiológicas y clínicas de pacientes que presentan en algún momento de su evolución POCS, describir la respuesta a distintos tratamientos y estudiar factores predictores de su evolución.
MétodosEstudio observacional retrospectivo de pacientes pediátricos con POCS seguidos en un hospital terciario en el periodo de noviembre 1997 a noviembre 2017.
ResultadosObtuvimos una muestra de 25 pacientes (68% varones). El 76% presentaba de base alteraciones en pruebas de neuroimagen o retraso psicomotor (POCS secundaria). El 24% restante eran niños sanos o con epilepsias benignas (POCS primaria). La edad media al inicio de la POCS fue de 6,7 años, siendo menor en los casos secundarios. Durante el periodo de POCS, el 72% presentó alguna manifestación clínica añadida. Todos recibieron combinaciones de antiepilépticos, siendo eficaces en el 36%. La POCS cesó en el 72%, siendo más probable el cese cuanto más tarde se hubiera instaurado. Un tercio tuvo alguna secuela, principalmente alteraciones cognitivas y del comportamiento, más frecuentes en POCS secundaria y en los pacientes en que duró más tiempo.
ConclusionesEl trazado electroencefalográfico de POCS, aunque infrecuente, supone un reto terapéutico. Es importante seguir a los pacientes con epilepsia, especialmente si asocia deterioro neurológico, para detectar la presencia POCS e iniciar tratamiento precoz.
Continuous spikes and waves during slow sleep (CSWS) is an EEG pattern that appears during childhood, and is often associated with cognitive impairment. It can appear in the course of epileptic syndromes, as well as in benign epilepsy. The aim of this study is to analyse epidemiological and clinical characteristic of patients with CSWS, in order to describe possible predictive factors in their outcome.
MethodsA retrospective study was conducted on paediatric patients with CSWS treated in a third-level hospital from November 1997 to November 2017.
ResultsThe study included 25 patients (68% male), of whom 76% had abnormalities in the neuroimaging or suffered from psychomotor development disorder (secondary CSWS). The rest were healthy, or diagnosed with idiopathic epilepsy. The mean age of onset of CSWS was 6.7 years, but earlier in the secondary CSWS cases. Symptoms were present during the CSWS episode in 72% of cases. All of them were treated with antiepileptic drugs, which were effective in 36%. CSWS stopped in 72%, and remission was longer if the CSWS onset occurred at an older age. One-third (33%) presented with sequelae, mostly cognitive and behavioural alterations. Outcome was poorer in those with secondary CSWS and, in those whose CSWS started at an earlier age and lasted longer.
ConclusionThe CSWS pattern, although rare, is still a therapeutic challenge. A close follow-up of the patients with epilepsy is important, especially if associated with cognitive impairment, in order to establish an early diagnosis and treatment.
En 1971, Patry et al.1 describieron por primera vez los casos de 6 niños que presentaban un patrón electroencefalográfico (EEG) de puntas y ondas difusas que ocupaban más del 85% del sueño no REM, asociado a deterioro cognitivo global. Lo llamaron «estado epiléptico eléctrico». Siete años después, Tassinari et al.2 describieron nuevos casos y propusieron que el estado epiléptico eléctrico durante el sueño (ESES, en sus siglas en inglés) era el responsable de las alteraciones neuropsicológicas de estos pacientes.
El término de punta-onda continua durante el sueño lento (POCS) fue introducido por la Liga Internacional contra la Epilepsia (ILAE) en 19893 y se ha utilizado indistintamente a ESES. La encefalopatía con POCS describe a pacientes con un trazado EEG típico de complejos de punta-onda bilaterales (a veces unilaterales) principalmente a 1,5-2,5Hz durante las fases del sueño lento (o no REM) (fig. 1), y una sintomatología caracterizada por deterioro cognitivo y crisis epilépticas4–6. Se presenta en niños de entre 2 y 14 años, con un pico de incidencia entre los 4 y 8 años7, y con mayor frecuencia descrita en varones6,8.
, sueño REM (b) y sueño no REM (c). Se objetivan durante el sueño no REM descargas de puntas y ondas de forma sincrónica sobre ambos hemisferios de forma continua, compatibles con POCS. Se ha registrado en el EEG la equivalencia a 1seg. Características del EEG: sensibilidad 10μV/mm; HF 70Hz; TC 0,3.")
Cambios en el EEG. Muestra fragmentos de un EEG de siesta de la misma paciente en vigilia (a), sueño REM (b) y sueño no REM (c). Se objetivan durante el sueño no REM descargas de puntas y ondas de forma sincrónica sobre ambos hemisferios de forma continua, compatibles con POCS. Se ha registrado en el EEG la equivalencia a 1seg. Características del EEG: sensibilidad 10μV/mm; HF 70Hz; TC 0,3.
El patrón EEG de POCS puede presentarse en otros síndromes epilépticos9 como el síndrome de Landau-Kleffner (que se caracteriza por una afasia adquirida asociada o no a crisis epilépticas)10, la epilepsia parcial benigna atípica de la infancia (formas poco frecuentes de epilepsia benigna que asocian crisis atónicas, crisis rolándicas nocturnas y a veces ausencias, con deterioro conductual y cognitivo)11,12 y la epilepsia benigna con puntas centrotemporales (epilepsia focal con crisis rolándicas nocturnas de pronóstico benigno, que excepcionalmente en su evolución puede desarrollar una POCS) (fig. 2).

Crisis durante POCS. Registro en el que se objetiva un episodio compatible con crisis epiléptica consistente en un cambio en el trazado que se sustituye por una punta-onda bilateral más irregular que la previamente registrada, sugiriendo que el patrón POCS se trata de un trazado intercrítico que se altera con la aparición de crisis epilépticas. Desde el punto de vista clínico la crisis consistió en parpadeos repetidos y clonismos de la musculatura peribucal. Se ha registrado en el EEG la equivalencia a 1seg. Características del EEG: sensibilidad 10μV/mm; HF 70Hz; TC 0,3.
Además el patrón de POCS puede aparecer en pacientes con daño cerebral previo (el 25% de los casos de POCS en algunas series5), siendo algunas de las más frecuentes: polimicrogiria, hidrocefalia y lesiones talámicas13,14.
El diagnóstico de POCS se realiza habitualmente mediante valoración directa del EEG de sueño, aunque algunos autores postulan el uso de «Spike-Wave Index», un parámetro cuantitativo definido como la suma de minutos que presentan punta-onda multiplicado por 100 y dividido por los minutos totales de sueño no REM2. Está muy discutido en la literatura el porcentaje necesario para ser considerado POCS. Clásicamente se propuso un Spike-Wave Index≥85%1, pero estudios posteriores han aceptado porcentajes menores: 50%8,13,15,16, 30%14 y hasta 25%17.
La etiopatogenia de las alteraciones neuropsicológicas asociadas a POCS no está clara; se piensa que la activación epiléptica anómala y mantenida durante el sueño interfiere con los procesos neuropsicológicos que suceden en el mismo, y con ello en la maduración cerebral, provocando una afectación grave de las funciones neurocognitivas y conductuales8,16,18,19. Para explicarlo con una analogía, Tassinari et al. en 200920 propusieron el nombre del «síndrome de Penélope», porque como en el mito de Penélope, mujer de Ulises, lo que tejía durante el día, era deshecho por la noche.
Algunos autores describen 3 periodos clínicos: un periodo inicial, antes de la POCS, en el que pueden existir crisis epilépticas; un periodo en el que la POCS está establecida en el que además de convulsiones se puede producir deterioro neurocognitivo, y un último periodo de secuelas.
Dentro de las alteraciones neuropsicológicas que se producen durante el periodo de POCS, los pacientes pueden presentar una regresión de funciones ya aprendidas e imposibilidad de un desarrollo posterior normal, pudiendo afectar a cualquier función superior (cognitiva, comportamiento, lenguaje, memoria, orientación) e incluso se han descrito alteraciones psicóticas6,21. También puede existir un deterioro en habilidades motoras previamente adquiridas.
El pronóstico de estos pacientes viene determinado por los déficits neuropsicológicos. El trazado EEG y las crisis epilépticas tienden a desaparecer, pero la mayoría de los pacientes mantienen alteraciones de las funciones cognitivas aun después de haber cesado la POCS5,6.
Por ello, el tratamiento está orientado a reducir la actividad EEG para no solo controlar las crisis, sino frenar el deterioro. Los tratamientos propuestos incluyen: fármacos antiepilépticos, corticoides, inmunoglobulinas, dieta cetogénica, estimulación del nervio vago e intervenciones quirúrgicas22. Las últimas revisiones indican que el tratamiento con corticoides y las resecciones quirúrgicas (en los casos con indicación) podrían ser alternativas más efectivas que los fármacos antiepilépticos7,23,24.
El objetivo de nuestro estudio es analizar las características epidemiológicas y clínicas previas y posteriores al desarrollo del patrón EEG de POCS de los niños seguidos en nuestro centro, así como estudiar posibles factores determinantes o variables pronósticas de su evolución clínica.
Sujetos y métodosRealizamos un estudio observacional retrospectivo de pacientes pediátricos con POCS seguidos en un hospital terciario (Hospital General Universitario Gregorio Marañón) desde noviembre de 1997 hasta noviembre de 2017. El estudio fue aprobado por el Comité de Ética de Investigación Clínica del hospital.
Se incluyeron todos los pacientes que habían presentado o presentaban en el momento de recogida de datos un patrón EEG POCS tratados en la sección de Neuropediatría del Hospital General Universitario Gregorio Marañón. Se recogieron los datos clínicos, se revisaron los registros video-EEG y los estudios de neuroimagen. Consideramos diagnóstico de POCS la presencia de complejos de punta-onda continua generalizada o focal en>85% del sueño no REM de un registro video-EEG prolongado.
Se recogieron un total 60 variables: demográficas y clínicas en los 3 periodos de estudio: antes del inicio de POCS, durante la fase de POCS, tras el cese de POCS o el fin del estudio.
En base a los resultados de los estudios de neuroimagen y la presencia o no de retraso psicomotor antes del inicio de la actividad EEG de POCS, se clasificaron los casos en:
- –
Secundarios: aquellos pacientes con alteraciones estructurales significativas en estudios de neuroimagen o aquellos que presentaban un retraso psicomotor previo. El retraso psicomotor fue medido con escalas de desarrollo ajustadas a la edad.
- –
No secundarios: aquellos con estudios de neuroimagen normales y sin retraso psicomotor antes del establecimiento de la POCS. Se incluyeron en este grupo los pacientes con síndrome de Landau-Kleffner, con epilepsia benigna atípica y con epilepsia idiopática que en su evolución presentasen POCS.
Las variables cuantitativas con distribución normal se expresaron en forma de media±desviación típica, mientras que las que no seguían una distribución normal se presentaron como mediana y rango intercuartílico (p25-p75). Las variables cualitativas se expresaron en forma de frecuencia y porcentajes. Se consideró estadísticamente significativa una p<0,05.
Se empleó el test Chi-cuadrado para el análisis de variables cualitativas o el test exacto de Fisher cuando fue necesario. Para el estudio de asociaciones estadísticas de variables cuantitativas se utilizó el análisis simple o bivariante: se usó el test de la t de Student para la comparación de variables con distribución normal entre grupos independientes o el test de U de Mann-Whitney si no seguían una distribución normal.
ResultadosHistoria previa al desarrollo de punta-onda continua durante el sueño lentoSe obtuvo una muestra de 25 pacientes formada por 17 varones y 8 mujeres, cuyo principal motivo de consulta fue crisis epilépticas (n=11, 44%), seguido de retraso psicomotor (n=6, 24%). La mediana de edad de la primera consulta fue de 1,9 años (0,4-5,5).
Cuatro pacientes presentaban enfermedades genéticas: síndrome de Wolf-Hirschhorn (deleción 4p), síndrome Down (trisomía 21), encefalopatía epiléptica secundaria a mutación del gen CASK y mutación gen FOXG1. Quince pacientes (60%) presentaban retraso psicomotor.
Veintiún pacientes (84%) tenían un registro EEG anterior al inicio de POCS, alterado en todos los casos y 9 de ellos presentaban actividad epiléptica en región rolándica.
Durante el tiempo de seguimiento se realizó resonancia magnética cerebral a todos los pacientes, encontrándose alteraciones en 15 casos (60%), entre las que destacan: atrofia cerebral (n=4); lesiones vasculares, siendo los infartos de la ACM lo más frecuente (n=4); y malformaciones cerebrales (n=10), entre las que destacan: polimicrogiria (n=3), hipogenesia/agenesia de cuerpo calloso (n=3), Chiari tipo 2 (n=1), encefalocele (n=1), hipoplasia de hipocampo (n=1) e hipoplasia de vérmix cerebeloso (n=1).
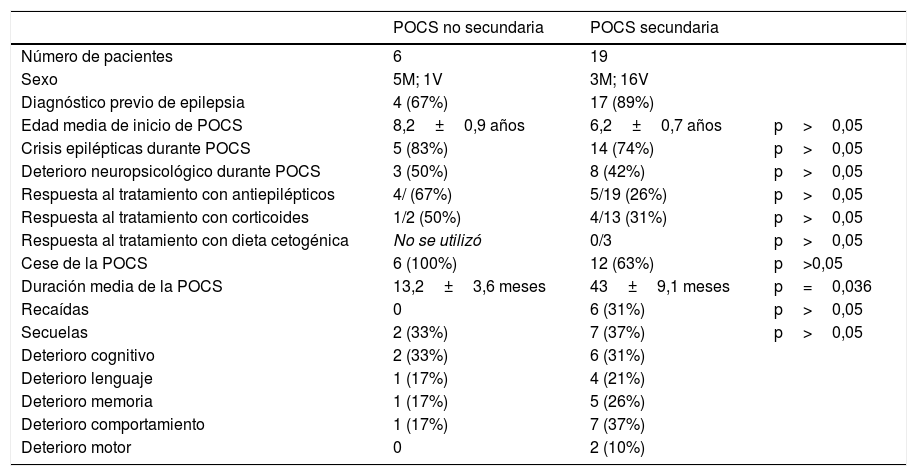
Periodo de punta-onda continua durante el sueño lentoSegún se ha descrito, diferenciamos 2 tipos de POCS en nuestro estudio: secundaria/sintomática/no idiopática (n=19) versus no secundaria/idiopática (n=6). Las características generales de estos pacientes se muestran en la tabla 1.
Comparación de POCS no secundaria versus POCS secundaria en cuanto a: características epidemiológicas y clínicas, inicio y cese de POCS, respuesta al tratamiento y secuelas
| POCS no secundaria | POCS secundaria | ||
|---|---|---|---|
| Número de pacientes | 6 | 19 | |
| Sexo | 5M; 1V | 3M; 16V | |
| Diagnóstico previo de epilepsia | 4 (67%) | 17 (89%) | |
| Edad media de inicio de POCS | 8,2±0,9 años | 6,2±0,7 años | p>0,05 |
| Crisis epilépticas durante POCS | 5 (83%) | 14 (74%) | p>0,05 |
| Deterioro neuropsicológico durante POCS | 3 (50%) | 8 (42%) | p>0,05 |
| Respuesta al tratamiento con antiepilépticos | 4/ (67%) | 5/19 (26%) | p>0,05 |
| Respuesta al tratamiento con corticoides | 1/2 (50%) | 4/13 (31%) | p>0,05 |
| Respuesta al tratamiento con dieta cetogénica | No se utilizó | 0/3 | p>0,05 |
| Cese de la POCS | 6 (100%) | 12 (63%) | p>0,05 |
| Duración media de la POCS | 13,2±3,6 meses | 43±9,1 meses | p=0,036 |
| Recaídas | 0 | 6 (31%) | p>0,05 |
| Secuelas | 2 (33%) | 7 (37%) | p>0,05 |
| Deterioro cognitivo | 2 (33%) | 6 (31%) | |
| Deterioro lenguaje | 1 (17%) | 4 (21%) | |
| Deterioro memoria | 1 (17%) | 5 (26%) | |
| Deterioro comportamiento | 1 (17%) | 7 (37%) | |
| Deterioro motor | 0 | 2 (10%) |
La edad media de inicio de POCS en el conjunto de la muestra fue de 6,7±2,9 años, siendo menor en varones (5,8±0,7 vs. 8.6±0,9; p=0,027) y en POCS secundaria (6,2±0,7 vs. 8,2±0,9 años, p=0,069). La frecuencia por grupo de edades se muestra en la figura 3.

La mediana del tiempo desde la primera crisis epiléptica, en los 21 pacientes que las padecían previamente, hasta el primer registro con patrón POCS fue de 27,7 meses (14,1-54,2).
En cuanto a las manifestaciones clínicas durante el periodo de POCS, 18 niños (72%) presentaron empeoramiento neurológico en comparación con su situación basal, medido de forma observacional. De los restantes 7 pacientes, en los que la única alteración fue el patrón EEG, 5 pertenecían al grupo de POCS secundaria y eran pacientes con retraso psicomotor moderado-grave por lo que fue difícil apreciar el deterioro neurológico.
Las principales manifestaciones clínicas fueron: crisis epilépticas (n=19; 76%), deterioro del lenguaje (n=8; 32%), alteraciones del comportamiento (n=8; 32%), alteraciones cognitivas (n=9; 36%), deterioro de las habilidades motoras previas (n=4;16%). Los datos de frecuencias de las distintas manifestaciones según el grupo de POCS se representan en la figura 4. No encontramos diferencias significativas entre los grupos.

Las crisis en este periodo fueron predominantemente generalizadas o parciales secundariamente generalizadas (80%), parciales simples (32%) y parciales complejas (20%). En el EEG, la POCS aparecía de forma bilateral difusa en 13 casos (52%) aunque con mayor actividad en el lado derecho en 9 (36%).
Se emplearon fármacos antiepilépticos en terapia combinada en todos los pacientes, siendo eficaces en 9 de ellos (36%). Entre los fármacos más utilizados se encuentran: ácido valproico (n=22; 88%), levetiracetam (n=18; 72%), clobazam (n=11; 44%) y etosuximida (n=7; 28%). Se emplearon 3 fármacos en 7 casos y hasta 5 fármacos de manera secuencial en 6 pacientes. Otros tratamientos empleados fueron: corticoterapia (n=15; 60%), siendo eficaz en 5 de ellos; y dieta cetogénica (n=3), sin objetivar mejoría. Consideramos eficaz el cese del trazado POCS. No se encontraron diferencias estadísticamente significativas entre la respuesta al tratamiento del grupo de POCS secundaria y no secundaria.
Cese de punta-onda continua durante el sueño lentoLa mediana de duración de la POCS en el conjunto de la muestra fue de 25,8 meses (10,9-41,7). La duración fue mayor en los casos secundarios que en los no secundarios (43±9,1 vs. 13,2±3,6 meses; p=0,036). La POCS cesó en 18 pacientes (72%), 12 de los cuales pertenecían al grupo de POCS secundaria y los otros 6 al de no secundarias, lo que supone el cese del 100% de los casos de POCS no secundaria frente al 63% de la secundaria (p=0,08).
Las crisis epilépticas remitieron en 12 de los 19 pacientes (63%) que las presentaron durante el periodo de POCS. Los 7 que mantenían crisis al final del estudio eran todos varones (p=0,03) y pertenecían al grupo de POCS secundaria (p=0,08).
Encontramos diferencias estadísticamente significativas en que los pacientes con mayor edad de inicio de POCS (por encima de los 6 años) presentaban más remisión del patrón EEG que aquellos que habían iniciado la POCS antes de los 6 años (p=0,029). Se consideró remisión como el cese del trazado EEG.
Durante la evolución, 6 pacientes presentaron una recaída, entendiendo como tal la reaparición del patrón POCS en el EEG cuando se había corregido previamente, y todos ellos pertenecían al grupo de POCS secundaria (p>0,05). Hubo más recaídas en los pacientes con más duración de POCS (p=0,30). La media de tiempo libre de POCS hasta la recaída fue de 16,8±11,6 meses. Cabe destacar que de los 18 pacientes libres de POCS al final del estudio, 2 habían presentado una recaída y de los 7 pacientes con persistencia de POCS, 4 habían pasado un periodo libre de dicho trazado de forma transitoria.
EvoluciónLos pacientes fueron seguidos una media de 8,4±4,6 años. Al final del estudio, la edad media de los pacientes era de 11,5±3,8 años y 9 niños (36%) presentaban secuelas, entendiendo por ello el empeoramiento con respecto a la situación previa al patrón de POCS. Las principales secuelas se muestran en la figura 5. De los 9 pacientes con secuelas, 7 pertenecían al grupo de POCS secundaria y 2 al no secundaria (p>0,05). Las diferencias en la frecuencia de secuelas entre los 2 grupos se muestran en la tabla 1.

Encontramos que las secuelas eran más frecuentes en pacientes en los que la POCS duró más y se inició de manera más precoz (p>0,05).
DiscusiónEl patrón EEG de POCS puede aparecer en pacientes con alguna patología de base, bien sean malformaciones, lesiones adquiridas cerebrales o retraso psicomotor previo, o en niños sanos o con diagnóstico de epilepsias benignas25. Al primer grupo nos referimos como POCS secundaria/sintomática/no idiopática y al segundo como POCS no secundaria.
Aunque la POCS suele asociarse a un deterioro neurológico, en ocasiones puede aparecer sin apreciarse deterioro cognitivo, en especial en pacientes con encefalopatías previas graves en los que evaluar el deterioro es más difícil. En nuestra serie encontramos más proporción de POCS secundaria que lo descrito clásicamente5, y mayor a lo referido en el metaanálisis de van den Munckhof23, lo que podría explicarse por las características de nuestro centro: un hospital de tercer nivel que atiende población con riesgo neurológico alto, como son los grandes prematuros o los pacientes con cardiopatías congénitas que pueden desarrollar lesiones cerebrales isquémicas. Si bien están descritos antecedentes familiares de epilepsia5, solo 2 niños en nuestra serie los presentaban, siendo este porcentaje menor a lo descrito.
Parece evidente que en la mayor parte de los casos, la POCS está precedida de una «fase de inicio» en la que los pacientes pueden presentar crisis epilépticas. El tipo de crisis descritas con mayor frecuencia son de tipo focal simple19 y en estudios más recientes se ha descrito actividad epiléptica en la región rolándica15. En nuestra serie el 84% pacientes presentaron crisis epilépticas antes del desarrollo de POCS, si bien lo más frecuente fueron crisis focales y generalizadas.
A la fase de inicio le sucede la «fase de estado», en la que aparece el patrón de POCS en el EEG pudiendo presentarse alteraciones neurocognitivas y crisis epilépticas. En nuestro estudio, el tiempo medio desde la primera crisis hasta el desarrollo de POCS es similar al descrito previamente, alrededor de 3 años16,21.
En nuestra serie el 76% de los niños presentó crisis epilépticas en la «fase de estado», similar a lo referido en la bibliografía26, constituyendo la manifestación clínica más frecuente. La mayoría de nuestros pacientes presentó durante este periodo crisis generalizadas, aunque se ha descrito la presencia de crisis de parciales o generalizadas4, y es frecuente la asociación de varios tipos de crisis6. Es característica la ausencia de crisis tónicas, a diferencia del síndrome de Lennox-Gastaut4,13.
Casi la mitad de los pacientes de nuestro estudio presentó deterioro neurocognitivo durante el periodo de POCS. El 52% restante no presentó deterioro respecto a su situación basal, lo que supone un porcentaje mayor que el descrito en la bibliografía. Esto se podría explicar por el alto porcentaje de pacientes con POCS secundaria de nuestra serie, muchos de ellos afectos de un retraso psicomotor grave en los que es difícil detectar un deterioro neurocognitivo añadido. La edad de inicio de POCS fue menor en el grupo sintomático, aunque no fue estadísticamente significativo, probablemente por tratarse de una muestra pequeña. Sin embargo, estudios con mayor tamaño muestral también describen un inicio más precoz en los casos sintomáticos14,25.
Durante la «fase de estado» se debe intensificar el tratamiento e intentar conseguir la remisión del trazado de POCS, ya que su duración se ha relacionado con peor pronóstico neurocognitivo5,7. Estudios recientes indican que el tratamiento con corticoides podría ser más efectivo para evitar el deterioro cognitivo7,23 y otros estudios muestran buenos resultados con el empleo de etosuximida en monoterapia o combinada con otros antiepilépticos, en especial con ácido valproico27,28. Sin embargo, como muestran los resultados, en nuestro estudio la respuesta a las diferentes modalidades de tratamiento fue menor a lo descrito en la literatura. Esto podría explicarse por 2 motivos: en primer lugar, por haber sido poco agresivos en el tratamiento de los pacientes en los que no se había apreciado deterioro neuropsicológico (que es un porcentaje alto de la serie); y, en segundo lugar, por ser la POCS secundaria más prevalente en nuestro estudio y tener menor remisión21.
Poco hay documentado sobre las recurrencias de POCS, pero en nuestro estudio la etiología (POCS secundaria/sintomática versus no secundaria/idiopática) resultó determinante en la evolución hacia el cese o recidiva de POCS ya que, aunque no se encontraron diferencias significativas, la POCS cesó en todos los casos no secundarios y todos ellos presentaron remisión de las crisis epilépticas, resultados similares a la serie de Caraballo et al.14,24. Una cuarta parte de nuestros pacientes presentaron una recidiva de POCS, todos ellos pertenecían al grupo de POCS sintomático.
La edad al inicio de POCS se presenta en nuestro estudio como un factor predictor de remisión, en el sentido de que a mayor edad de inicio (por encima de 6 años) se produce más remisión del trazado. Esto podría estar en relación con que la actividad epiléptica mantenida en edades críticas del neurodesarrollo (más tempranas) puede alterar la maduración cerebral y producir una afectación evolutiva grave. Diferentes estudios apoyan estos resultados y muestran peor evolución cuando el inicio de la POCS se produce antes de los 6 años29.
La evolución de los pacientes con POCS es variable en las distintas series. La duración de la POCS también es variable14,21, estando relacionada con el pronóstico neurológico como apoya la literatura5,7,30, lo que también se observa en nuestro estudio: se producen más secuelas y recaídas en aquellos en los que la POCS duró más y se inició de forma más precoz, aunque en nuestro estudio esta diferencia no fue significativa, probablemente por el pequeño tamaño muestral.
En cuanto a la «fase de secuelas», un tercio de nuestra muestra presenta alguna secuela neurocognitiva al final del seguimiento, más frecuentes en el grupo POCS secundaria, sin ser la diferencia significativa. En comparación con otros estudios, nuestro porcentaje de secuelas ha sido bajo, probablemente porque como secuela hemos considerado un empeoramiento con respecto a la situación de base y, al incluir más pacientes con retraso psicomotor previo, el deterioro poco evidente puede haberse infradiagnosticado. Otra explicación sería que los pacientes con POCS sintomática tuvieron menos manifestaciones neurocognitivas durante la «fase de estado», por lo que es más probable que ello haya derivado en menos secuelas.
Entre las limitaciones de nuestro estudio señalamos el pequeño tamaño muestral, el diseño retrospectivo y, en especial, la falta de test neuropsicológicos que valoren el desarrollo de forma objetiva, ya que somos conscientes de las limitaciones de extraer conclusiones de valoraciones subjetivas. Sin embargo, los especialistas responsables son profesionales habituados a valorar el desarrollo psicomotor en la infancia
ConclusionesNuestro estudio apoya la idea de que los pacientes con POCS deben ser diagnosticados y tratados, especialmente si asocian deterioro cognitivo, con un seguimiento estrecho dado el impacto que tiene en su desarrollo neurológico.
Recomendamos el seguimiento y reconocimiento precoz de las señales de alarma que indiquen el inicio de POCS en los pacientes diagnosticados previamente de epilepsia. Es importante un diagnóstico y tratamiento precoz para que el patrón EEG de POCS persista el menor tiempo posible.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
