





Continuous spikes and waves during slow sleep (CSWS) is an EEG pattern that appears during childhood, and is often associated with cognitive impairment. It can appear in the course of epileptic syndromes, as well as in benign epilepsy. The aim of this study is to analyse epidemiological and clinical characteristics of patients with CSWS, in order to describe possible predictive factors in their outcome.
MethodsA retrospective study was conducted on paediatric patients with CSWS treated in a third-level hospital from November 1997 to November 2017.
ResultsThe study included 25 patients (68% male), of whom 76% had abnormalities in the neuroimaging or suffered from psychomotor development disorder (secondary CSWS). The rest were healthy, or diagnosed with idiopathic epilepsy. The mean age of onset of CSWS was 6.7 years, but earlier in the secondary CSWS cases. Symptoms were present during the CSWS episode in 72% of cases. All of them were treated with antiepileptic drugs, which were effective in 36%. CSWS stopped in 72%, and remission was longer if the CSWS onset occurred at an older age. One-third (33%) presented with sequelae, mostly cognitive and behavioural alterations. Outcome was poorer in those with secondary CSWS and, in those whose CSWS started at an earlier age and lasted longer.
ConclusionThe CSWS pattern, although rare, is still a therapeutic challenge. A close follow-up of the patients with epilepsy is important, especially if associated with cognitive impairment, in order to establish an early diagnosis and treatment.
La punta-onda continua durante el sueño lento (POCS) es un trazado electroencefalográfico característico, que aparece en la infancia y que en ocasiones condiciona un deterioro cognitivo. Este patrón electroencefalográfico puede aparecer tanto en determinados síndromes epilépticos como en la evolución de epilepsias idiopáticas y sintomáticas. El objetivo de nuestro estudio es analizar las características epidemiológicas y clínicas de pacientes que presentan en algún momento de su evolución POCS, describir la respuesta a distintos tratamientos y estudiar factores predictores de su evolución.
MétodosEstudio observacional retrospectivo de pacientes pediátricos con POCS seguidos en un hospital terciario en el periodo de noviembre 1997 a noviembre 2017.
ResultadosObtuvimos una muestra de 25 pacientes (68% varones). El 76% presentaba de base alteraciones en pruebas de neuroimagen o retraso psicomotor (POCS secundaria). El 24% restante eran niños sanos o con epilepsias benignas (POCS primaria). La edad media al inicio de la POCS fue de 6,7 años, siendo menor en los casos secundarios. Durante el periodo de POCS, el 72% presentó alguna manifestación clínica añadida. Todos recibieron combinaciones de antiepilépticos, siendo eficaces en el 36%. La POCS cesó en el 72%, siendo más probable el cese cuanto más tarde se hubiera instaurado. Un tercio tuvo alguna secuela, principalmente alteraciones cognitivas y del comportamiento, más frecuentes en POCS secundaria y en los pacientes en que duró más tiempo.
ConclusionesEl trazado electroencefalográfico de POCS, aunque infrecuente, supone un reto terapéutico. Es importante seguir a los pacientes con epilepsia, especialmente si asocia deterioro neurológico, para detectar la presencia POCS e iniciar tratamiento precoz.
In 1971, Patry et al.1 were the first to describe 6 cases in children of an electroencephalographic (EEG) diffuse spike-wave pattern that spanned over more than 85% of non-rapid eye movement (non-REM) sleep and was associated with global cognitive deterioration. They labelled it “electrical status epilepticus”. Seven years later, Tassinari et al.2 described additional cases and proposed that electrical status epilepticus during sleep (ESES) was responsible for the neuropsychological abnormalities observed in these patients.
The term continuous spike and waves during slow–wave sleep (CSWS) was first introduced by the International League Against Epilepsy (ILAE) in 19893 and has been used interchangeably with ESES. Encephalopathy with CSWS refers to patients with a typical EEG pattern of bilateral (sometimes unilateral) spike-wave discharges (mainly at 1.5–2.5Hz) during slow–wave (non-REM) sleep (Fig. 1) and a clinical picture characterised by cognitive deterioration and epileptic seizures.4–6 It manifests in children aged 2–14 years, with an incidence peak at ages 4–8 years,7 and occurs more frequently in males.6,8
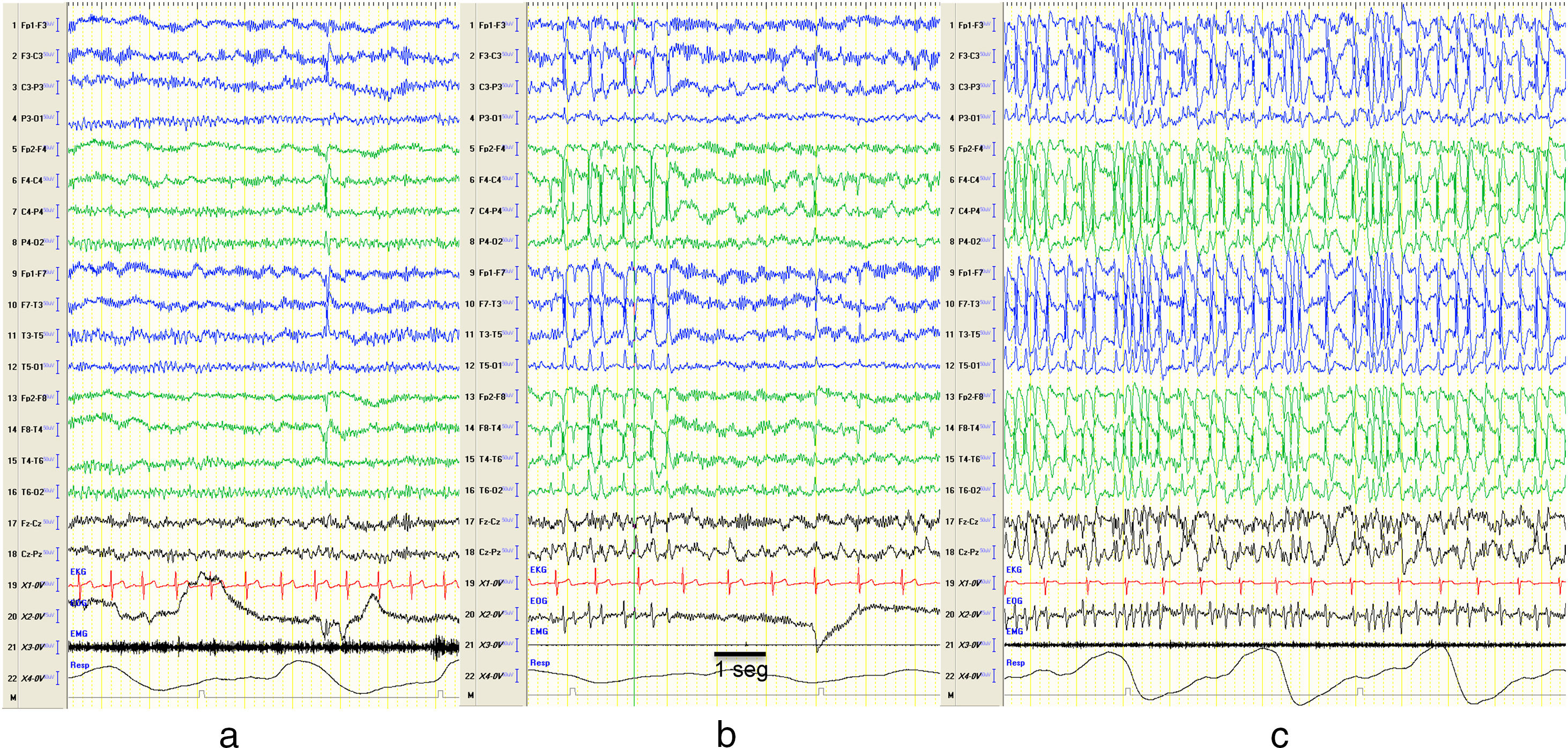
Changes in EEG patterns. Example of portions of an EEG recording during naptime in a patient while awake (a), in REM sleep (b) and non-REM sleep (c). Presence of synchronic and continuous spike and wave discharges on both hemispheres compatible with CSWS during non-REM sleep. The horizontal line drawn at the bottom shows the length equivalent to 1s. EEG machine settings: sensitivity 10μV/mm; HF 70Hz; TC 0.3.
The EEG pattern of CSWS may manifest in the context of other epilepsy syndromes,9 such as Landau-Kleffner syndrome (characterised by acquired aphasia presenting with or without epileptic seizures),10 atypical benign partial epilepsy of childhood (infrequent forms of benign epilepsy manifesting with atonic seizures, nocturnal sylvian seizures and sometimes absences, behavioural problems and cognitive deterioration)11,12 and benign epilepsy of childhood with centrotemporal spikes (focal epilepsy with benign sylvian seizures at night, in the context of which CSWS may develop in exceptional cases) (Fig. 2).
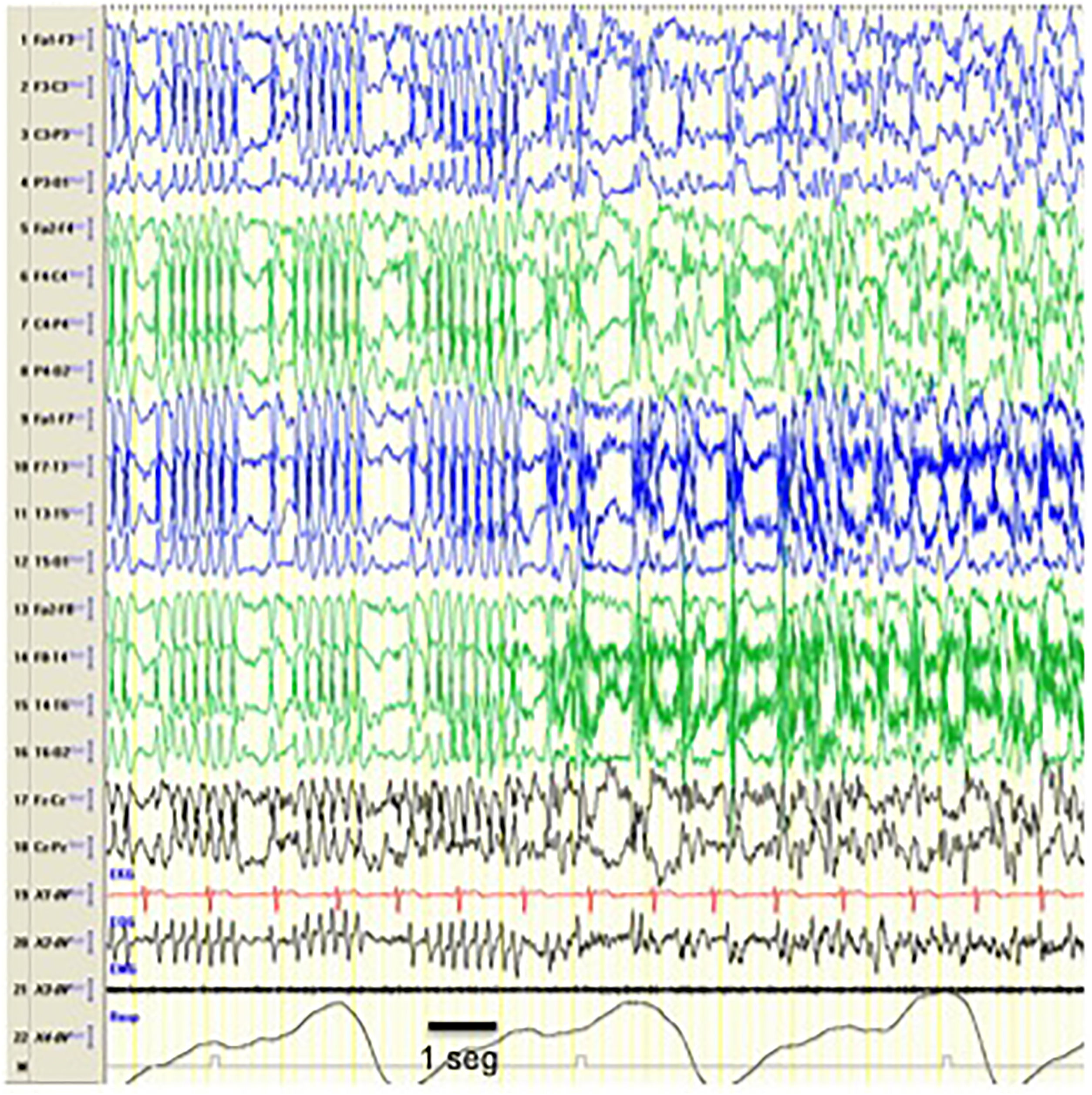
Seizure during CSWS. Tracings showing an episode compatible with an epileptic seizure with a change in the pattern, which is replaced by bilateral spike-wave discharges that are more irregular compared to the previously recorded pattern, suggesting that CSWS is an interseizure pattern that is altered by the development of epileptic seizures. From a clinical standpoint, the seizure manifested with repetitive blinking and jerking movements of the peribuccal muscles. The horizontal line drawn at the bottom shows the length equivalent to 1 second. EEG machine settings: sensitivity 10μV/mm; HF 70Hz; TC 0.3.
The CSWS pattern can also manifest in patients with a history of brain damage (25% of cases of CSWS in some case series5), and some of the most frequent associated conditions are polymicrogyria, hydrocephalus and thalamic lesions.13,14
The diagnosis of CSWS is usually made by direct assessment by means of a sleep EEG, although some authors have proposed the use of the Spike-Wave Index, a quantitative parameter defined as the sum of all spike and slow wave minutes multiplied by 100 and divided by the total non-REM sleep minutes.2 There is wide disagreement in the literature as to which is the minimum percentage required to define CSWS using this index. Historically, authors proposed a Spike-Wave Index of at least 85%,1 but more recent studies have considered smaller percentages acceptable: 50%,8,13,15,16 30%14 and even 25%.17
The pathophysiology underlying the neuropsychological abnormalities associated with CSWS has not been established; it has been hypothesised that the sustained abnormal epileptic activity during sleep interferes with the neuropsychological processes that occur during this state and therefore with brain maturation, resulting in serious disturbances of neurocognitive functioning and behaviour.8,16,18,19 To illustrate this hypothesis, Tassinari et al. proposed an analogy in 2009,20 labelling the process “Penelope syndrome” since, as in the myth of Penelope, the wife of Odysseus, in this disease what is woven during the day is unravelled during the night.
Some authors have described 3 electroclinical stages: a prodromal stage, preceding onset of CSWS, during which the patient may experience epileptic seizures; an acute stage during which CSWS manifests and seizures may be accompanied by neurocognitive deterioration, and a residual stage after cessation of CSWS characterised by its sequelae.
Among the neuropsychological abnormalities that manifest during the acute stage of CSWS, patients may experience regression of functions that had been previously acquired, which may preclude normal development in the future, with potential impairment of any of the higher-order functions (cognition, behaviour, language, memory, orientation) and in some cases development of psychotic features.6,21 Patients may also experience impairment of previously acquired motor skills.
Outcomes in these patients are determined by the resulting neuropsychological deficits. The characteristic EEG pattern and epileptic seizures usually disappear, but most patients continue to have cognitive impairments after CSWS has resolved.5,6
For this reason, the treatment is aimed at reducing the epileptiform activity evinced by EEG not only with the aim of controlling seizures, but also to halt deterioration. The treatments proposed for this purpose include antiepileptic drugs, steroid therapy, immunoglobulins, a ketogenic diet, vagal nerve stimulation and surgical intervention.22 According to the most recent systematic reviews, steroid therapy and surgical resection (where indicated) could be more effective than antiepileptic drugs.7,23,24
The aim of our study was to analyse the epidemiological and clinical characteristics of patients before and after the development of the CSWS EEG pattern in children managed in our hospital, and to assess potential determinants or predictors of patient outcomes.
Sample and methodsWe conducted a retrospective observational study in paediatric patients with CSWS managed in a tertiary care hospital (Hospital General Universitario Gregorio Marañón) from November 1997 to November 2017. The study was approved by the Clinical Research Ethics Committee of the hospital.
We included all patients managed in the Department of Paediatric Neurology of the Hospital General Universitario Gregorio Marañón that had exhibited or were currently exhibiting an EEG pattern of CSWS. We collected clinical data and reviewed video-EEG recordings and neuroimaging tests. We defined CSWS as the presence of continuous focal or generalised spike and wave discharges during 85% of slow–wave sleep in a prolonged video-EEG recording.
We collected data for 60 demographic and clinical variables for the 3 stages under study (prodromal, acute and residual) or through the end of the follow-up period.
Based on the findings of neuroimaging tests and the presence or absence of psychomotor retardation before the EEG onset of CSWS, we classified cases into:
- –
Secondary: patients with significant structural abnormalities identified in the imaging tests or with pre-existing psychomotor retardation. We measured psychomotor retardation using age-appropriate developmental scales.
- –
Nonsecondary: patients with normal neuroimaging findings and without psychomotor retardation prior to onset of CSWS. This group included patients with Landau-Kleffner syndrome, atypical benign epilepsy and idiopathic epilepsy that developed CSWS at any point during their follow-up.
We have summarised quantitative variables as mean±standard deviation if they followed a normal distribution, and as median and interquartile range (IQR) otherwise. We have summarised qualitative variables as absolute frequencies and percentages. We defined statistical significance as p- normally distributed value of less than 0.05.
We analysed qualitative data by means of the chi square test or the Fisher exact test as applicable. We assessed the association between quantitative variables by means of univariate and bivariate analyses, using the Student's t test to compare normally distributed variables and the Mann–Whitney U test otherwise.
ResultsHistory prior to the onset of continuous spike and waves during slow sleepWe obtained a sample of 25 patients (17 male and 8 female) in whom the most frequent initial presenting complaint had been epileptic seizures (n=11; 44%), followed by psychomotor retardation (n=6; 24%). The median age at the time of the first visit was 1.9 years (IQR, 0.4–5.5).
Four patients had genetic disorders: Wolf-Hirschhorn disease (chromosome 4p deletion), Down syndrome (trisomy 21), epileptic encephalopathy secondary to a CASK gene and a FOXG1 mutation. Fifteen patients (60%) had psychomotor retardation.
Twenty-one patients (84%) had exhibited epileptiform EEG patterns prior to onset of CSWS, abnormal in every case, with epileptiform activity in the centrotemporal area in 9.
All patients underwent a head MRI scan during the follow-up, with abnormal findings in 15 cases (60%), chief of which were cerebral atrophy (n=4); vascular lesions, most frequently middle cerebral artery stroke (n=4); and cerebral malformations (n=10) including polymicrogyria (n=3), hypoplasia/agenesis of the corpus callosum (n=3), Chiari malformation type 2 (n=1), encephalocoele (n=1), hippocampal hypoplasia (n=1) and cerebellar vermis hypoplasia (n=1).
Acute stage of continuous spike and wave during slow sleepAs stated above, we differentiated between 2 types of CSWS in our study: secondary/symptomatic/non-idiopathic (n=19) versus nonsecondary/idiopathic (n=6). Table 1 presents the general characteristics of these patients.
Comparison of nonsecondary CSWS and secondary CSWS in terms of: epidemiological and clinical characteristics, onset and cessation of CSWS, response to treatment and sequelae.
| Nonsecondary CSWS | Secondary CSWS | ||
|---|---|---|---|
| Number of patients | 6 | 19 | |
| Sex | 5F, 1M | 3M; 16V | |
| Previous diagnosis of epilepsy | 4 (67%) | 17 (89%) | |
| Mean age at onset of CSWS | 8.2±0.9 years | 6.2±0.7 years | P>.05 |
| Epileptic seizures during CSWS | 5 (83%) | 14 (74%) | P>.05 |
| Neuropsychological deterioration during CSWS | 3 (50%) | 8 (42%) | P>.05 |
| Response to antiepileptic drugs | 4/ (67%) | 5/19 (26%) | P>.05 |
| Response to steroid therapy | 1/2 (50%) | 4/13 (31%) | P>.05 |
| Response to ketogenic diet | Not used | 0/3 | P>.05 |
| Cessation of CSWS | 6 (100%) | 12 (63%) | P>.05 |
| Mean duration of CSWS | 13.2±3.6 months | 43±9.1 months | P=.036 |
| Relapses | 0 | 6 (31%) | P>.05 |
| Sequelae | 2 (33%) | 7 (37%) | P>.05 |
| Cognitive impairment | 2 (33%) | 6 (31%) | |
| Language impairment | 1 (17%) | 4 (21%) | |
| Memory impairment | 1 (17%) | 5 (26%) | |
| Behavioural impairment | 1 (17%) | 7 (37%) | |
| Motor impairment | 0 | 2 (10%) |
F, female; M, male.
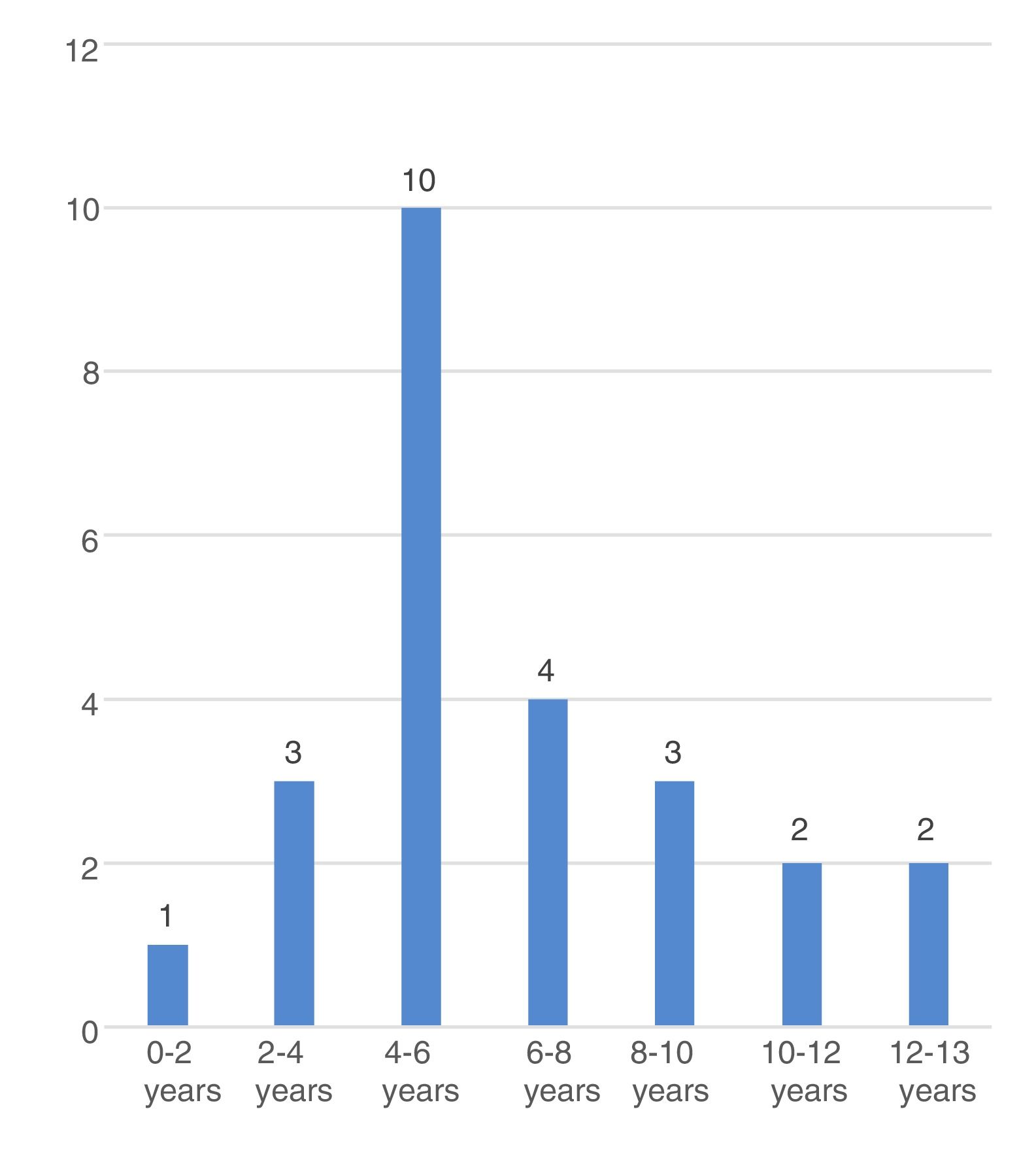
The mean age at onset of CSWS in the overall sample was 6.7±2.9 years, with a younger age in male patients (5.8±0.7 vs. 8.6±0.9; P=.027) and in patients with secondary CSWS (6.2±0.7 vs. 8.2±0.9 years, P=.069). Fig. 3 shows the distribution of each time by age group.
The median time elapsed from the first epileptic seizure to the first EEG recording with a CSWS pattern in the 21 patients that had a previous history of epilepsy was 27.7 months (IQR, 14.1–54.2).
When it came to the manifestations during the acute stage of CSWS, 18 children (72%) exhibited neurologic worsening compared to baseline, which was assessed by subjective observation. Of the remaining 7 patients, in whom the only change was in the EEG pattern, 5 belonged to the secondary CSWS group and had moderate to severe psychomotor retardation, due to which it was difficult to assess any ensuing neurologic deterioration.
The most frequent clinical manifestations were epileptic seizures (n=19; 76%), language impairment (n=8; 32%), changes in behaviour (n=8; 32%), cognitive changes (n=9; 36%) and deterioration of previously acquired motor skills (n=4; 16%). Fig. 4 presents the frequency of the different clinical manifestations by type of CSWS. We did not find any differences between groups.
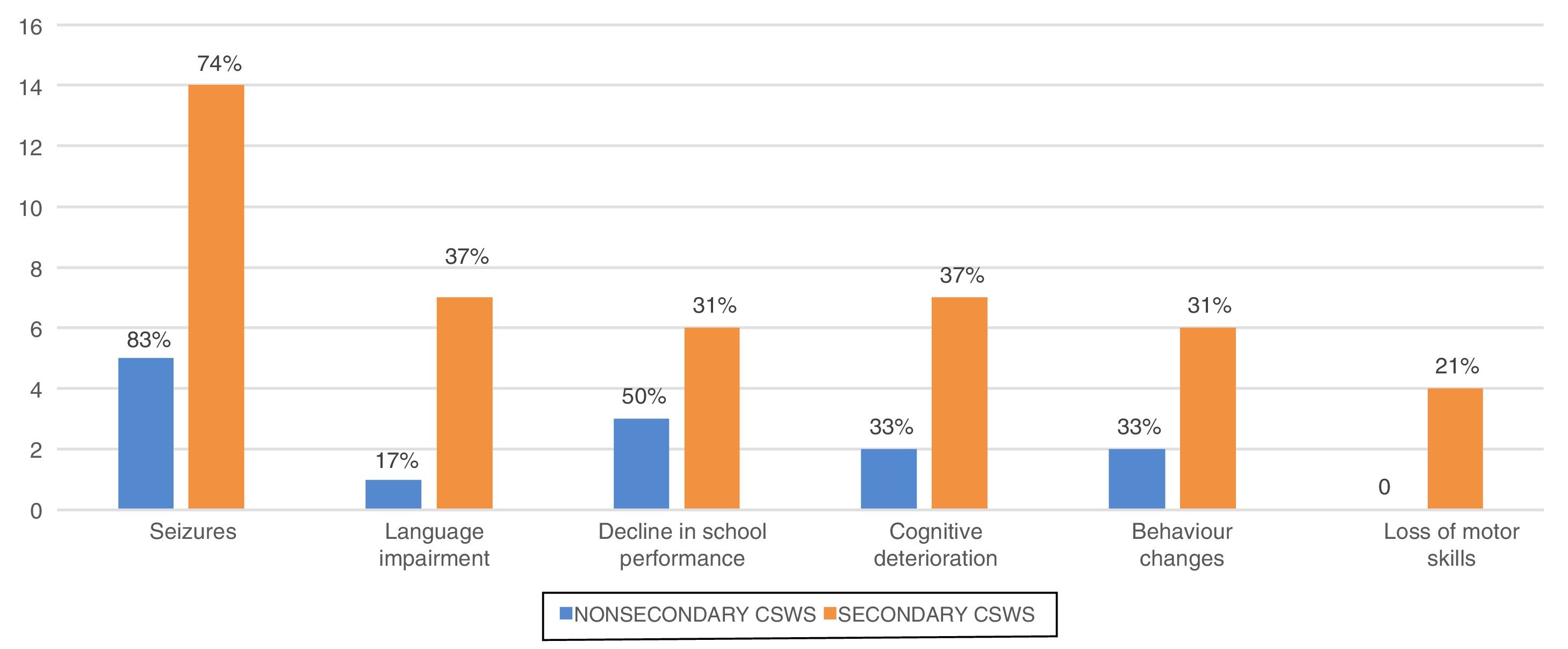
During this period, the most frequent seizures were generalised or secondary generalised seizures (80%) followed by simple partial seizures (32%) and complex partial seizures (20%). In EEG recordings, CSWS was bilateral and diffused in 13 cases (52%), although 9 exhibited a predominance of activity in the right (36%).
Combination therapy with antiepileptic drugs was used in all patients and effective in 9 (36%). The most frequently used drugs were valproic acid (n=22; 88%), levetiracetam (n=18; 72%), clobazam (n=11; 44%) and ethosuximide (n=7; 28%). Three drugs were used in 7 cases, and up to 5 drugs were used sequentially in 6 patients. Other treatments used in these patients were steroid therapy (n=15; 60%), which was effective in 5 patients, and the ketogenic diet (n=3), with no evidence of improvement in any. We defined treatment response as cessation of CSWS in EEG tracings. We did not find statistically significant differences in the response to treatment between the secondary and the nonsecondary CSWS groups.
Cessation of continuous spike and wave during slow sleepThe median duration of CSWS in the overall sample was of 25.8 months (IQR, 10.9–41.7). The duration was longer in the secondary CSWS group compared to the nonsecondary CSWS group (43±9.1 vs 13.2±3.6 months; P=.036). Cessation of CSWS occurred in 18 patients (72%), 12 in the secondary CSWS group and 6 in the nonsecondary CSWS group, corresponding to cessation in 100% of nonsecondary cases compared to 63% of secondary cases (P=.08).
Epileptic seizures remitted in 12 of the 19 patients (63%) that experienced during the acute stage of CSWS. The 7 patients that continued to have seizures by the end of the follow-up period were all male (P=.03) and belonged to the secondary CSWS group (P=.08).
We found a statistically significant difference in remission by age of onset, with remission of the EEG pattern being more frequent in patients with onset at older ages (age>6 years) compared to patients with onset of CSWS before age 6 years (P=.029). We defined remission as disappearance of the CSWS EEG pattern.
During the follow-up, 6 patients experienced recurrences, understood as the reappearance of the CSWS pattern on EEG tracings after a previous remission, and all of these patients belonged to the secondary CSWS group (P>.05). Recurrences were more frequent in patients with longer duration of CSWS (P=.30). The mean time free of CSWS until recurrence was of 16.8±11.6 months. We ought to highlight that of the 18 patients free of CSWS by the end of the study, 2 had experienced a recurrence, while of the 7 patients with persistent CSWS, 4 had experienced a transient period free of the CSWS EEG pattern.
OutcomesThe mean duration of follow-up in our sample was 8.4±4.6 years. At the end of data collection, the mean age of the patients was 11.5±3.8 years and 9 children (36%) had sequelae, which we defined as being in worse clinical condition compared to the condition before the onset of the EEG CSWS pattern. Fig. 5 presents the most frequent sequelae. Of the 9 patients with sequelae, 7 belonged to the secondary CSWS group and 2 to the nonsecondary CSWS group (P>.05). Table 1 shows the differences in the distribution of sequelae between the 2 groups.
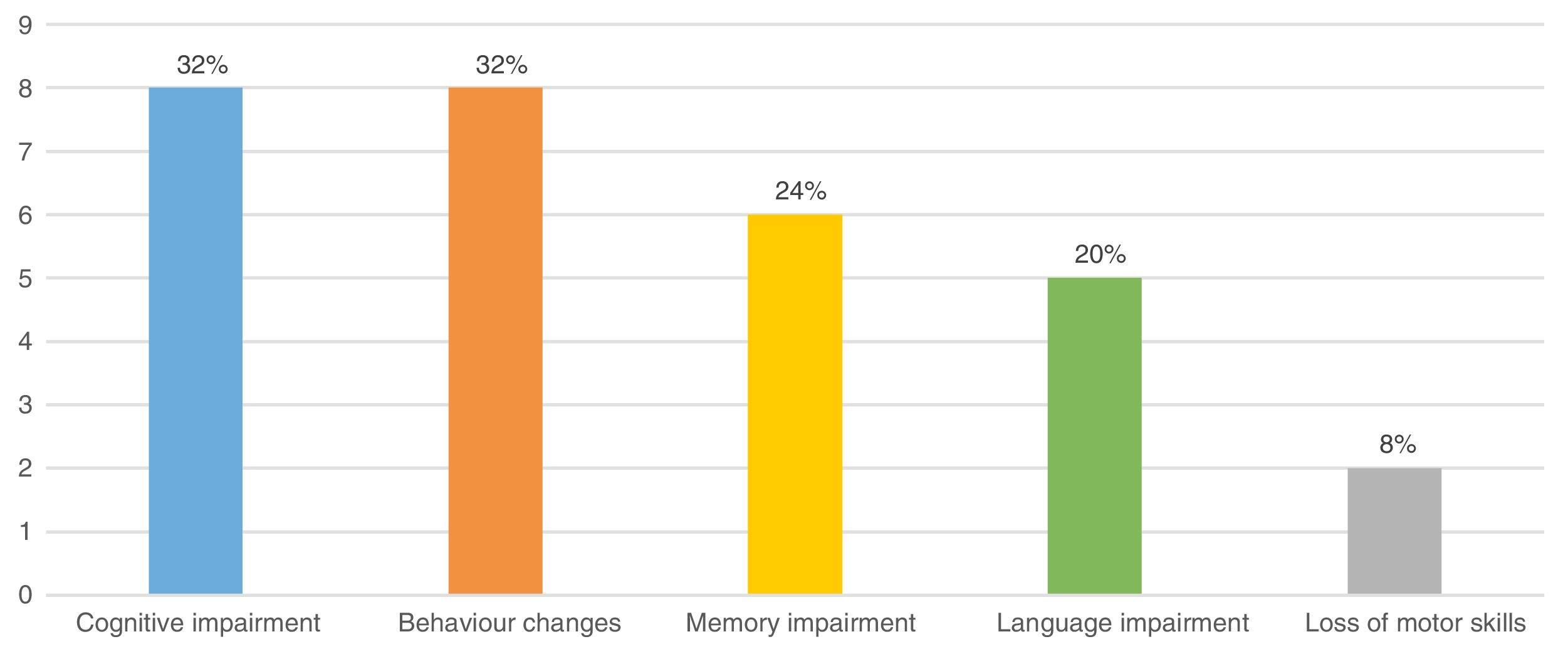
We found a higher frequency of sequelae in patients with earlier onset and longer duration of CSWS (P>.05).
DiscussionThe EEG pattern of CSWS may appear in patients with underlying disorders, such as congenital malformations, acquired brain damage or pre-existing psychomotor retardation, or in children previously healthy or with a diagnosis of benign epilepsy.25 We refer to the same group as secondary/symptomatic/non-idiopathic CSWS and the second group as nonsecondary CSWS.
Although CSWS is usually associated with neurologic impairment, there are cases with no appreciable cognitive deterioration, especially in patients with a previous history of severe encephalopathy in whom assessment of deterioration is more challenging. In our case series, we found a higher proportion of secondary CSWS than previously described5 and found in the meta-analysis by van den Munckhof,23 which could be explained by the characteristics of our study setting: a tertiary care referral hospital serving populations with a high risk of neurologic disease, such as patients born very preterm or with congenital heart defects that may lead to development of ischaemic brain lesions. Although a family history of epilepsy has been described in the previous literature,5 we only found it in 2 children in our sample, corresponding to a lower proportion.
It appears that in most cases, the onset of CSWS is preceded by a “prodromal stage” during which patients may exhibit epileptic seizures. The type of seizures reported most frequently in this period are simple focal seizures,19 and more recent studies have described epileptiform activity in the centrotemporal region.15 In our sample, 84% of patients had epileptic seizures prior to onset of CSWS, although the most frequent type were focal to generalised seizures.
This prodromal stage is followed by an “acute stage” characterised by the development of the CSWS EEG pattern, possibly associated with neurocognitive changes and epileptic seizures. In our study, the mean time elapsed from the first epileptic seizure to onset of CSWS was similar to the one described in previous studies, of approximately 3 years.16,21
In our series, 76% of children had epileptic seizures during the “acute stage”, which was consistent with the previous literature,26 and this was the most frequent clinical manifestation. Most patients in our sample experienced generalised seizures during this stage, although other authors have reported the presence of partial or generalised seizures4 and a combination of different types of seizures is also frequent.6 Contrary to the presentation of Lennox-Gastaut syndrome, tonic seizures are typically absent in cases of CSWS.4,13
Nearly half of the patients in our study exhibited neurocognitive deterioration during the acute stage of CSWS. The remaining 52% did not exhibit apparent deterioration compared to baseline, a percentage that was higher compared to previous reports in the literature. This may be due to the high percentage of patients with secondary CSWS in our series, many of who had severe psychomotor retardation prior to onset and in whom it was therefore difficult to discern any additional neurocognitive impairment. The age at onset of CSWS was lower in the symptomatic group, although this difference was not statistically significant, probably due to the small sample size. Nevertheless, studies with larger samples have also described an earlier onset in symptomatic patients.14,25
During the “acute stage”, treatment must be intensified in an attempt to achieve remission of the CSWS EEG pattern, its duration is directly associated with poorer neurocognitive outcomes.5,7 Some recent studies suggest that steroid therapy may be more effective in preventing cognitive deterioration,7,23 while others have reported favourable outcomes with the use of ethosuximide as monotherapy or in combination with other antiepileptic drugs, especially valproic acid.27,28 However, as can be seen in the results section of this article, in our study the response to different treatment modalities was lesser compared to previous descriptions in the literature. We can think of 2 possible reasons: first, because treatment of patients with no detectable neurologic worsening (who amounted to a large proportion of the sample) was not as aggressive as it could have been, and second, that secondary CSWS was more prevalent in our study and is associated with a lower frequency of remission.21
The published data on the recurrence of CSWS is scarce, but in our study, the aetiology of CSWS (secondary/symptomatic CSWS vs nonsecondary/idiopathic CSWS) seemed to be a determinant of cessation of CSWS or relapse, for, while the differences we found were not significant, CSWS resolved and was accompanied by remission of epileptic seizures in all nonsecondary cases, which was similar to the findings of the case series published by Caraballo et al.14,24 One fourth of our patients experienced recurrence of CSWS, and all belonged to the symptomatic CSWS group.
In our study, the age at onset of CSWS was a predictor of remission, by which we mean that an older age at onset (age>6 years) was associated with a higher frequency of EEG remission. A possible explanation is that epileptic activity at ages that are critical for neurodevelopment (younger ages) may alter cerebral maturation and result in serious abnormalities in development. Various studies have corroborated these findings, with evidence of poorer outcomes in patients with onset of CSWS before age 6 years.29
The outcomes of patients with CSWS vary between case series. The reported duration of CSWS also varies,14,21 and the current evidence suggests that it is associated with the neurodevelopmental outcomes,5,7,30 which was also the case in our study: we found more sequelae and relapses in patients with earlier onset and longer duration of CSWS, although in our study these differences were not statistically significant, probably due to the small sample size.
When it came to the residual “sequelae” stage, 1/3 of our sample exhibited some form of neurocognitive deterioration by the end of the follow-up period, with a higher prevalence in patients in the secondary CSWS group, although this difference was not statistically significant. In our study, the proportion of patients with sequelae was smaller compared to previous studies, probably because we defined sequelae as a worsening compared to the baseline condition of the patient, and since our study included a larger proportion of patients with pre-existing psychomotor retardation, we may have underdiagnosed subtle additional impairments. Another possible explanation is that patients with symptomatic CSWS may have had fewer neurocognitive manifestations during the acute stage, which would have resulted in fewer sequelae.
Among the limitations of our study, we ought to highlight the small sample size, its retrospective design and, most importantly, that development was not objectively assessed with neuropsychological tests, as we are aware of the limitations of conclusions drawn from subjective assessments. However, it should also be taken into account that the specialists involved in the study are professionals that routinely assess psychomotor development in children in their clinical practice.
ConclusionsOur findings suggest that patients with CSWS need to receive an accurate diagnosis and appropriate treatment, and, especially if they experience cognitive impairment, remain under close monitoring given the significant impact of this disease on neurodevelopment.
We recommend close follow-up of patients with a previous diagnosis of epilepsy and a high level of suspicion for the early detection of signs alerting of the possible onset of CSWS. Early diagnosis and treatment are essential to minimise the duration of the EEG pattern of CSWS.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Escobar Fernández L, Coccolo Góngora A, Vázquez López M, Polo Arrondo AP, Miranda Herrero MC, Barredo Valderrama E, et al. Patrón punta-onda continua en el sueño lento: nuestra experiencia durante 20 años. An Pediatr (Barc). 2019;91:180–188.
