








El síndrome de hipoventilación central congénita (SHCC) es una enfermedad genética muy rara causada por mutaciones en PHOX2B; en 2010 se creó el Consorcio Europeo del Síndrome de Hipoventilación Central, que en 2012 implantó un Registro online de pacientes para optimizar su cuidado.
ObjetivoConocer las características y la evolución de los pacientes españoles con SHCC y detectar áreas de mejora.
Materiales y métodoSe analizaron los datos actualizados en diciembre del 2015 de los pacientes españoles del Registro europeo.
ResultadosSe registró a 38 pacientes, nacidos entre 1987 y 2013, procedentes de 18 hospitales. El 34,2% eran mayores de 18 años. Han fallecido 3 pacientes. Aportaban estudio del gen PHOX2B 37 (97,3%), 32 (86,5%) con mutación. Los genotipos 20/25, 20/26 y 20/27 representaron el 84,3% de las mutaciones. Las disautonomías fueron más frecuentes y graves en portadores de genotipos con mayores expansiones de polialaninas. El 47% de pacientes asociaba alteraciones oculares, el 16% Hirschsprung, el 13% hipoglucemias y el 5% tumores. Treinta pacientes (79%) debutaron en el periodo neonatal y 8 (21%) posteriormente (inicio/diagnóstico tardío). Ocho niños (21%) recibieron inicialmente ventilación domiciliaria con mascarilla; 5 eran lactantes con comienzo neonatal, 2 de ellos precisaron cambio a traqueostomía tras presentar parada cardiorrespiratoria; ambos tenían mutaciones graves. Han sido decanulados y transferidos a mascarilla el 34,3% de los pacientes (edad media: 13,7 años). El 29,4% de los niños escolarizados precisaron refuerzo educativo.
ConclusiónLa implementación del Registro en España de pacientes con SHCC ha permitido identificar aspectos relevantes para optimizar sus cuidados, tales como la importancia del estudio genético para el diagnóstico y la estimación de gravedad, la frecuencia elevada de alteraciones oculares y de necesidad de refuerzo educativo, y algunas limitaciones de las técnicas ventilatorias.
Congenital Central Hypoventilation Syndrome (CCHS) is a very rare genetic disease. In 2012 the European Central Hypoventilation Syndrome (EuCHS) Consortium created an online patient registry in order to improve care.
AimTo determine the characteristics and outcomes of Spanish patients with CCHS, and detect clinical areas for improvement.
Materials and methodAn assessment was made on the data from Spanish patients in the European Registry, updated on December 2015.
ResultsThe Registry contained 38 patients, born between 1987 and 2013, in 18 hospitals. Thirteen (34.2%) were older than 18 years. Three patients had died. Genetic analysis identified PHOX2B mutations in 32 (86.5%) out of 37 patients assessed. The 20/25, 20/26 and 20/27 polyalanine repeat mutations (PARMs) represented 84.3% of all mutations. Longer PARMs had more, as well as more severe, autonomic dysfunctions. Eye diseases were present in 47%, with 16% having Hirschsprung disease, 13% with hypoglycaemia, and 5% with tumours. Thirty patients (79%) required ventilation from the neonatal period onwards, and 8 (21%) later on in life (late onset/presentation). Eight children (21%) were using mask ventilation at the first home discharge. Five of them were infants with neonatal onset, two of them, both having a severe mutation, were switched to tracheostomy after cardiorespiratory arrest at home. Approximately one-third (34.3%) of patients were de-cannulated and switched to mask ventilation at a mean age of 13.7 years. Educational reinforcement was required in 29.4% of children attending school.
ConclusionThe implementation of the EuCHS Registry in Spain has identified some relevant issues for optimising healthcare, such as the importance of genetic study for diagnosis and assessment of severity, the high frequency of eye disease and educational reinforcement, as well as some limitations in ventilatory techniques.
El síndrome de hipoventilación central congénita (SHCC) o enfermedad de Ondine es una patología genética muy rara del sistema nervioso autónomo (SNA), caracterizada por la pérdida del control automático de la respiración1,2; se manifiesta típicamente durante el sueño no REM o, en casos más graves, en todo el sueño, incluso en vigilia. Puede asociar otras disautonomías (alteraciones del ritmo cardiaco3, de la motilidad intestinal y ocular4,5, enfermedad de Hirschsprung6 y tumores de la cresta neural7,8).
Se produce por mutaciones en heterocigosis del gen paired-like Homeobox 2B (PHOX2B), cuya función es clave en el desarrollo del SNA durante la embriogénesis9,10. Las mutaciones más frecuentes (90% de los casos), denominadas «poly-ala-repeat mutations» (PARM), consisten en expansiones del segmento de 20 residuos de alanina presente en el exón 3 de PHOX2B; la longitud de dichas expansiones es variable, habiéndose detectado genotipos desde 20/24 hasta 20/33. El 10% restante de las mutaciones identificadas, denominadas «non-poly-ala-repeat mutations» (NPARM), son mutaciones puntuales missense, pequeñas inserciones/deleciones que alteran la pauta de lectura11, o, más raramente (< 1%), deleciones/duplicaciones del exón 3 o del gen completo12. La mayoría de las mutaciones se producen de novo, aunque hasta un 25% de los progenitores asintomáticos pueden tener mosaicismo somático13. El estudio genético confirma el diagnóstico clínico e informa sobre la gravedad, ya que las mutaciones con expansiones más cortas (20/24 y 20/25) presentan penetrancia variable, pudiendo manifestarse incluso con fenotipo normal; por el contrario, las PARM con mayores expansiones (20/27 y 20/33) asocian disautonomías de mayor gravedad y mayor necesidad de ventilación mecánica (VM) 24 h14,15. Las mutaciones NPARM asocian mayor incidencia de tumores y enfermedad de Hirschsprung16.
Generalmente, la dependencia de VM aparece desde el nacimiento (SHCC de inicio precoz); no obstante, a veces la hipoventilación pasa desapercibida o se manifiesta más tarde, diagnosticándose después del mes de vida, incluso en adultos portadores de mutación (SHCC de inicio o diagnóstico tardío)17-20. Existen otras formas tardías de hipoventilación central primaria sin mutación conocida, entidades diferentes del SHCC, tales como la entidad “rapid-onset obesity with hypoventilation, hypothalamic dysfunction and autonomic dysregulation” (ROHHAD)21,22.
Los niños afectados de SHCC precisan óptima VM en domicilio para sobrevivir y/o alcanzar un óptimo neurodesarrollo, por lo que el diagnóstico precoz y la vigilancia continua de su estado respiratorio son claves para evitar complicaciones y secuelas derivadas de la hipoxemia y la hipercapnia crónica15,23.
Es una enfermedad muy rara, con incidencia desconocida, aunque estimada de 1/200.000 recién nacidos (RN) vivos24. En 2010 se organizó el Consorcio Europeo del Síndrome de Hipoventilación Central, para optimizar el cuidado de los pacientes25 (www.ichsnetwork.eu/); en este marco fue diseñado un Registro europeo online de pacientes, siguiendo las normas de seguridad, anonimicidad y confidencialidad vigentes. Catorce países europeos participan actualmente, incluida España. Los objetivos del presente trabajo son: a) conocer las características fenotípicas y genotípicas, formas de presentación, técnicas de VM utilizadas y evolución de los pacientes españoles diagnosticados de SHCC incluidos en el Registro europeo, y b) identificar aspectos relevantes en el cuidado de estos pacientes, subsidiarios de mejora.
MétodoLa implantación del Registro europeo en España fue autorizada por un Comité Ético de Investigación Clínica español de referencia y se divulgó a través de sociedades científicas y de la Asociación Ondine, que integra a familiares y pacientes españoles con SHCC (www.sindromedeondine.es). Se comenzó el reclutamiento de pacientes en marzo del 2012. Cuatro familias declinaron participar. Los responsables legales y/o pacientes firmaron el consentimiento informado para participar. Los médicos españoles responsables de los pacientes, uno de ellos coordinador nacional, recibieron acceso al Registro online; cada investigador introdujo los datos, obtenidos mediante revisión de la historia clínica y entrevista a los afectados, supervisados posteriormente por el coordinador. Los datos se actualizan periódicamente, la última fecha, el 31 de diciembre del 2015.
Se recogieron los siguientes datos: fecha y país de nacimiento, sexo, raza, datos del embarazo, parto y periodo neonatal. Manifestaciones clínicas iniciales y evolutivas (respiratorias, gastrointestinales, neurológicas, cardiovasculares, oculares, endocrinas, tumores, otras); patologías asociadas. Diagnóstico: fecha, pruebas realizadas, estudio genético, mutación encontrada. Tratamientos: técnicas de ventilación y cambios; cirugías. Tiempo de ingreso hospitalario. Aspectos sociales: escolarización, grado académico alcanzado, primer trabajo. Datos del fallecimiento.
Se trata de un estudio observacional transversal. La descripción de los datos se realizó mediante la media, la mediana, la desviación típica y el rango de las variables cuantitativas, así como mediante las frecuencias absoluta y relativa de las variables cualitativas. Cuando los datos lo permitieron, se realizaron contrastes de hipótesis; las medias de los días de ingreso se compararon mediante el test de Mann-Whitney y la relación entre la frecuencia de patologías asociadas y las diversas mutaciones se estudió mediante regresión lineal. Los datos relevantes se presentaron en tablas y figuras. Se utilizó el programa SPSS versión 15 para el análisis estadístico.
ResultadosDurante el periodo del estudio (desde marzo del 2012 hasta diciembre del 2015) se registraron y actualizaron los datos de 38 pacientes, nacidos entre 1987 y 2013, de edad media 13,6 ± 7,45 años; mediana 11,35 meses; rango de edad: 5 meses y 28,6 años. Trece pacientes (34,2%) eran mayores de 18 años. Diecisiete (44,7%) eran varones y 21 (55,3%) mujeres. La distribución de pacientes por comunidad autónoma de residencia fue la siguiente: Madrid 7 pacientes, Andalucía 6, Castilla-La Mancha 5, Cataluña 5, Murcia 3, País Vasco 3, Aragón 2, Valencia 2, Canarias 1, Castilla-León 1, Extremadura 1, Islas Baleares 1 y Navarra 1 paciente. En el estudio han participado 22 investigadores, pertenecientes a 19 hospitales españoles, referidos en la lista de autores.
Han fallecido 3 pacientes (7,8%), todos de forma inesperada fuera del hospital, 2 durante el sueño y otro por accidente; sus edades de defunción fueron 5 meses, 13 meses y 15 años, respectivamente.
Genotipos, fenotipos y correlación genotipo/fenotipoTreinta y siete pacientes aportaban estudio del gen PHOX2B, realizado en 32 niños (86,4%) en el Instituto de Genética Médica y Molecular del Hospital La Paz de Madrid, laboratorio de referencia en España para el diagnóstico de SHCC. Treinta y dos pacientes (86,4%) presentaban mutación; 30 mutaciones (93%) eran PARM y 2 (6,6%) eran NPARM (tabla 1). Los genotipos 20/25, 20/26 y 20/27 representan el 84,3% de las mutaciones encontradas. El estudio genético fue negativo en 4 niños: 2 estaban diagnosticados de síndrome de ROHHAD por el cuadro clínico asociado; uno inició hipoventilación a los 10 años; uno, con comienzo neonatal, presentaba hipoventilación en sueño tranquilo. Se desconoce el resultado del estudio en un paciente y no fue realizado en otro.
Correlación genotipo-fenotipo. Número y porcentaje (entre paréntesis) de pacientes que asocian las diversas patologías del sistema nervioso autónomo en cada grupo y subgrupo de mutación del gen PHOX2B (PARM —20/25, 20/26, 20/27, 20/33—, NPARM)
| 20/25 | 20/26 | 20/27 | 20/33 | Total PARM | NPARM | No mutación | NR | Total | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (n=6) | (n=10) | (n=11) | (n=3) | (n=30) | (%) | (n=2) | (%) | (n=5) | (n=1) | (n=38) | (%) | |
| Tumores SNA | – | – | – | – | – | 1 (50) | 1 | _ | 2 | (5,2) | ||
| Enfermedad de Hirschsprung | – | – | 3 | 2 | 5 | (16,6) | 1 (50) | _ | _ | 6 | (15,8) | |
| Vólvulo gastrointestinal | – | – | 1 | 1 | 2 | (6,6) | – | – | – | 2 | (5,2) | |
| Síncopes | – | 1 | 3 | – | 4 | (13,3) | – | – | – | 4 | (10,5) | |
| Mareos | 1 | 1 | 1 | 3 | (10) | – | – | – | 3 | (7,9) | ||
| Marcapasos cardiaco | – | 1 | 1 | – | 2 | (6,6) | – | – | – | 2 | (5,2) | |
| Convulsiones | 3 | 3 | 5 | 1 | 12 | (40) | – | 2 | – | 14 | (36,8) | |
| Espasmos del sollozo | – | – | 2 | 2 | 4 | (13,3) | – | 1 | – | 5 | (13,1) | |
| Hipoglucemias | 1 | 2 | 1 | 1 | 5 | (16,6) | – | – | – | 5 | (13,1) | |
| Alteraciones oculares | 1 | 4 | 9 | 3 | 17 | (56,6) | – | – | 1 | 18 | (47,3) | |
| Media n.° patologías/pac pa | 1 | 1,2 < 0,7 | 2,36 < 0,014 | 3,33 < 0,003 | ||||||||
| Media de días de estanciab p | 228 < 0,46 | 215 < 0,46 | 429 < 0,016 | 87 < 0,07 | 282 | 425 < 0,94 | 165 < 0,98 | 152 < 0,92 | 271 | |||
NR: estudio genético no realizado; NPARM: nonpolyalanine repeat expansion mutation; PARM: polyalanine repeat expansion mutation; SNA: sistema nervioso autónomo.
La tabla 1 muestra las patologías asociadas y su relación con las diferentes mutaciones; destacan las alteraciones oculares por su alta frecuencia (47,3%). Dos pacientes fueron intervenidos de urgencia por vólvulo gastrointestinal. Cinco pacientes (13,1%) presentaron hipoglucemias.
En esta serie no es posible comparar los tipos de patologías del SNA encontrados en ambos tipos de mutaciones (PARM vs. NPARM) porque solo 2 pacientes presentan mutación NPARM. Sin embargo, las patologías estructurales (tumores del SNA, Hirschsprung) tienden a asociarse con mutaciones NPARM y las patologías funcionales (alteración del ritmo cardiaco, hipoglucemias, etc.) se asocian con las mutaciones PARM (tabla 1).
Dentro de las mutaciones PARM, las patologías del SNA son más frecuentes y más graves en pacientes con expansiones mayores (20/27 y 20/33; p<0,014 y p<0,003, respectivamente) (tabla 1).
Formas clínicas de presentación inicialTreinta pacientes (79%) comenzaron de forma típica, necesitando VM desde el periodo neonatal (29 desde el primer día de vida, 1 después), sin patología cardiopulmonar u otra justificación (inicio precoz); de ellos, 28 (93,3%) presentaban mutación, uno no, y se desconoce en otro. Ocho pacientes (21%) (tabla 2) comenzaron con otros síntomas y/o la necesidad de VM se detectó o apareció en edad posterior (inicio-diagnóstico tardío), 4 de ellos (50%) presentaban mutación.
Pacientes con inicio o diagnóstico tardío del síndrome de hipoventilación central
| Síntomas previos | Edad de inicio VM | Síntomas al comienzo (modo de ventilación) | Mutación |
|---|---|---|---|
| 1. RN sano, alta a domicilioa | 19 días | Cianosis, hipercapnia sin distrés, necesidad de VM (TQ) | 20/26 |
| 2. RN sano, alta a domicilio | 40 días | Letargia, rechazo de tomas, cianosis, hipercapnia sin distrés respiratorio, necesidad de VM (TQ) | 20/25 |
| 3. RN 1.er ddv: episodios de cianosis; diagnóstico sepsis clínica, antibióticos, alta a domicilio | 46 días | Reingresa por cianosis, hipercapnia sin distrés, necesidad de VM (TQ) | 20/25 |
| 4. RN 1.er ddv: episodios de cianosis, hipoglucemias, sepsis clínica, antibióticos, alta a domicilio | 50 días | Reingresa por letargia, rechazo de tomas, cianosis, hipercapnia sin distrés respiratorio, edemas, hepatopatía, necesidad de VM (TQ) | 20/25 |
| 5. RN 3.er ddv síntomas digestivos, necesita NPT; un mes: hipercapnia no valorada; 3 meses: diagnosticado de enfermedad de Hirschsprung | 5,8 meses | Bronquiolitis, necesidad de VM (VNI) | 20/33 |
| 6b. Trastornos del sueño 14 meses: hipertensión pulmonar primaria | 5,6 años | Neumonía, hipoxemia, crisis convulsivas, necesidad de VM (TQ) | No realizado |
| 7b. Sano hasta 7 meses antes, cuando inicia polifagia, rápida ganancia de peso, enuresis, alteraciones del sueño, sudoración profusa | 3,9 años | Apnea, cianosis, PCR. ROHHAD: obesidad, hipoventilación central, disfunción hipotalámica, ganglioneuroblastoma torácico, necesidad de VM (TQ) | No mutación |
| 8. Sano hasta los 3 años, cuando inicia polifagia, gana 14kg en pocos meses, poliuria, sudoración profusa | 5,2 años | Apneas centrales y obstructivas. Bradicardias, Raynaud, ROHHAD: obesidad, hipoventilación central, disfunción hipotalámica (TSH elevada), necesidad de VM (VNI) | No mutación |
| 9. Sano | 10,4 años | Coma, hipercapnia, «encefalitis» resuelta con VM, RM normal, persiste hipoventilación central, necesidad de VM (VNI) | No mutación |
ddv: días de vida; E: enfermedad; NPT: nutrición parenteral total; PCR: parada cardiorrespiratoria; RM: resonancia magnética; RN: recién nacido; ROHHAD: rapid-onset obesity with hypoventilation, hypothalamic dysfunction and autonomic dysregulation; TQ: traqueostomía; VM: ventilación mecánica; VNI: ventilación no invasiva con mascarilla.
Técnicas ventilatorias en la Unidad de Cuidados Intensivos pediátrica o neonatal: Durante el comienzo de la enfermedad, 35 pacientes (92,1%) recibieron intubación y ventilación invasiva, y 18 (47%) ventilación no invasiva (VNI), usada por primera vez en el año 2000. Quince niños (39,5%) utilizaron ambos modos.
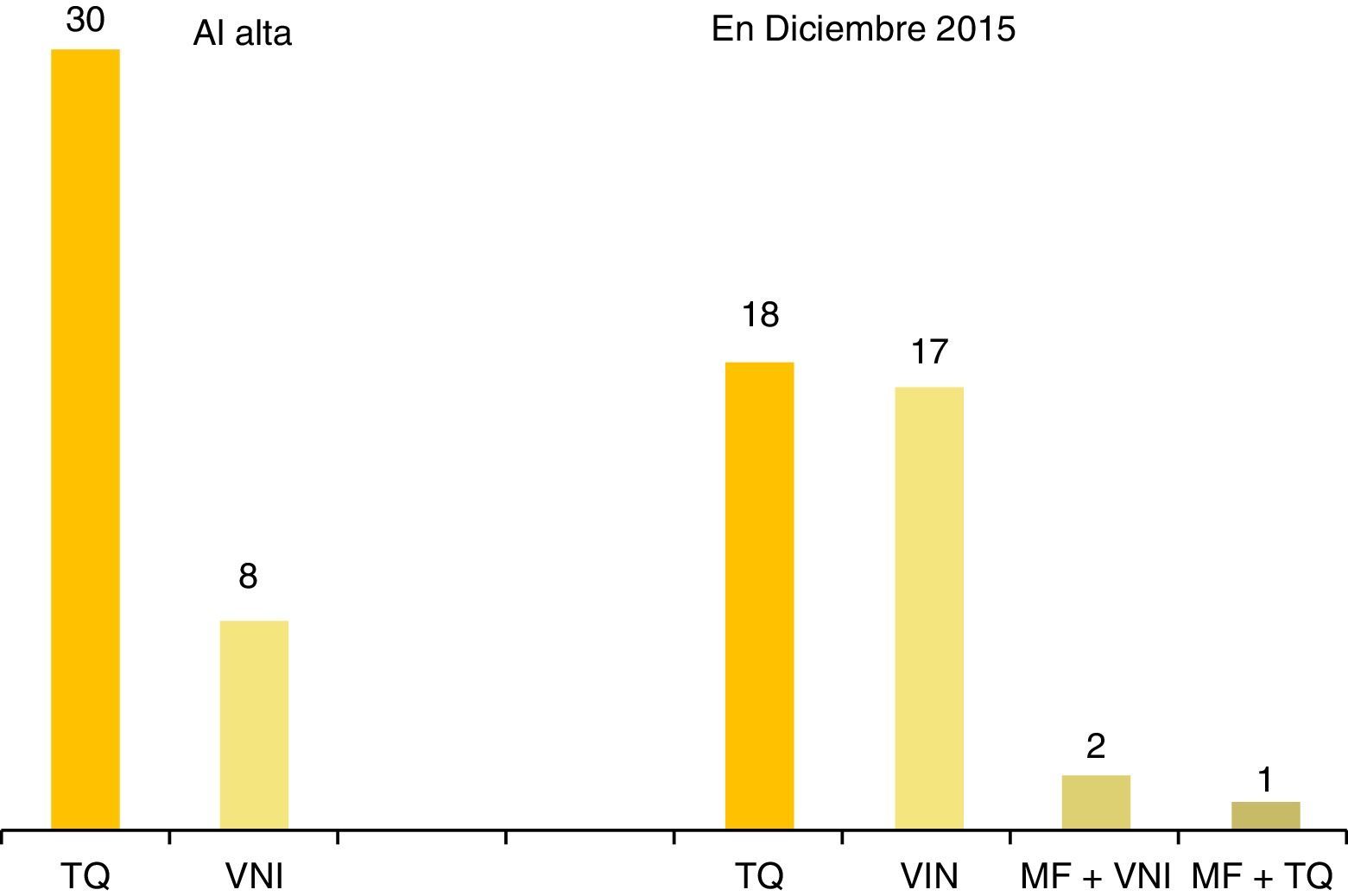
Técnicas ventilatorias en domicilio: a) traqueostomía (TQ): 32 pacientes (84,2%) han sido ventilados con TQ; 6 (15,8%) nunca fueron traqueostomizados; la edad media del procedimiento quirúrgico fue de 7,8 meses ± 1,1; mediana, 2,6 meses; rango 24 días-5,75 años. Han sido decanulados 11 pacientes (34,3%); la edad media de decanulación y transferencia a VNI fue de 13,7 años ± 1,89; mediana, 12,66 años; rango 6,12-20,5 años. Solo un paciente era menor de 10 años cuando fue decanulado. b) Ventilación con mascarilla: fue usada en domicilio por 19 pacientes (50%), la primera vez en 1998, evento publicado por sus autores26. La tabla 3 muestra las características de los 8 niños ventilados con mascarilla en el momento de su primer alta a domicilio tras el diagnóstico; 5 eran lactantes con comienzo neonatal de la enfermedad, 2 de los cuales precisaron cambio a TQ tras presentar parada cardiorrespiratoria en domicilio por ventilación ineficaz (genotipos: 20/33, NPARM). c) Marcapasos frénico (MF): se implantó en 4 pacientes (11%), en el Hospital de Parapléjicos de Toledo, a la edad/año de implantación de: 1,2 años/1992 (que fracasó), 8,8 años/2000, 23 años/2013 y 24 años/2014; los 3 pacientes ventilados con MF en diciembre del 2015 eran adultos dependientes de VM 24 h, que en vigilia usaban el MF y por la noche, la TQ (un paciente) o la mascarilla (2 pacientes).
Pacientes en los que la ventilación no invasiva con mascarilla se usó como primera opción y se mantuvo en el primer alta a domicilio
| Edad de inicio de ventilación | Edad de alta a domicilio | Patología asociada | Mutación | Cambio a otro modo | Edad de cambio | Motivo de cambio | |
|---|---|---|---|---|---|---|---|
| 1 | 1.er ddv | 2,7 meses | No | 20/25 | No | ||
| 2 | 1.er ddv | 2,17 meses | No | 20/25 | No | ||
| 3 | 1.er ddv | 1,7 meses | No | No mutación | No | ||
| 4 | 1.er ddv | 2,43 meses | Hirschsprung Espasmos del sollozo | 20/33 | Sí, a TQ | 5, 7 meses | PCR en casa |
| 5 | 1.er ddv | 2,23 meses | No | NPARM | Sí, a TQ | 1, 6 años | PCR en casa |
| 6 | 5,7 meses | 7,3 meses | Hirschsprung | 20/33 | No | ||
| 7 | 5,2 años | 5, 3 años | ROHHAD | No mutación | No | ||
| 8 | 10,4 años | 10,9 años | Neuropatía periférica | No mutación | Sí, asocia MF | 22,9 años | Necesidad de VM 24 h |
ddv: día de vida; MF: marcapasos frénico; NPARM: nonpolyalanine repeat expansion mutation; PCR: parada cardiorrespiratoria; ROHHAD: rapid-onset obesity with hypoventilation, hypothalamic dysfunction and autonomic dysregulation; TQ: traqueostomía; VM: ventilación mecánica.
A lo largo de su vida en domicilio, 22 pacientes han recibido un sólo modo de ventilación, 13, 2 modos, y 3, 3 modos. Ningún paciente fue ventilado con presión negativa. La figura 1 muestra las técnicas ventilatorias aplicadas en la primera alta a domicilio y en diciembre del 2015.
Estancias: en las tablas 1 y 4 se describe la duración del ingreso que originó la VM a domicilio. El tiempo fue significativamente más largo en pacientes con genotipo 20/27 (p<0,016), dados de alta con TQ (p<0,004) y en ingresados antes del año 2000 (p<0,000).
Duración (días) del ingreso en el que se inició la ventilación mecánica a domicilio. Fue significativamente mayor en los pacientes con TQ y en los ingresados antes del año 2000
| Todos | Modo de ventilación | Tipo de comienzo | Año de inicio del ingreso | ||||
|---|---|---|---|---|---|---|---|
| (n=38) | TQ (n=30 | VNI (n=8) | Neonatal (n=30) | Tardío (n=8) | <2000 (n=13) | ≥ 2000 (n=25) | |
| Media | 271 | 319* | 91 | 304a | 150 | 566b | 118 |
| Desviación típica | 313 | 336 | 51 | 344 | 69 | 392 | 53 |
| Mediana | 153 | 181 | 70 | 153 | 162 | 332 | 103 |
| Rango | 49-1165 | 62-1165 | 49-175 | 51-1165 | 49-259 | 154-1165 | 49-212 |
TQ: traqueostomía; VNI: ventilación no invasiva con mascarilla.
a. Escolarización: de los 34 pacientes en edad escolar, 3 (8,8%) recibieron educación especial y 31 (91%) acudieron a colegio normal, aunque 10 de estos (29,4%) necesitaron refuerzo educativo continuado. La figura 2 muestra el grado académico alcanzado según la edad, menor o mayor de 18 años. Cuatro pacientes mayores de 18 años (30%) recibieron educación superior (3, universidad y 1, grado formativo superior), b) trabajo: 4 pacientes mayores de 18 años (30%) iniciaron actividad laboral remunerada.
Discusión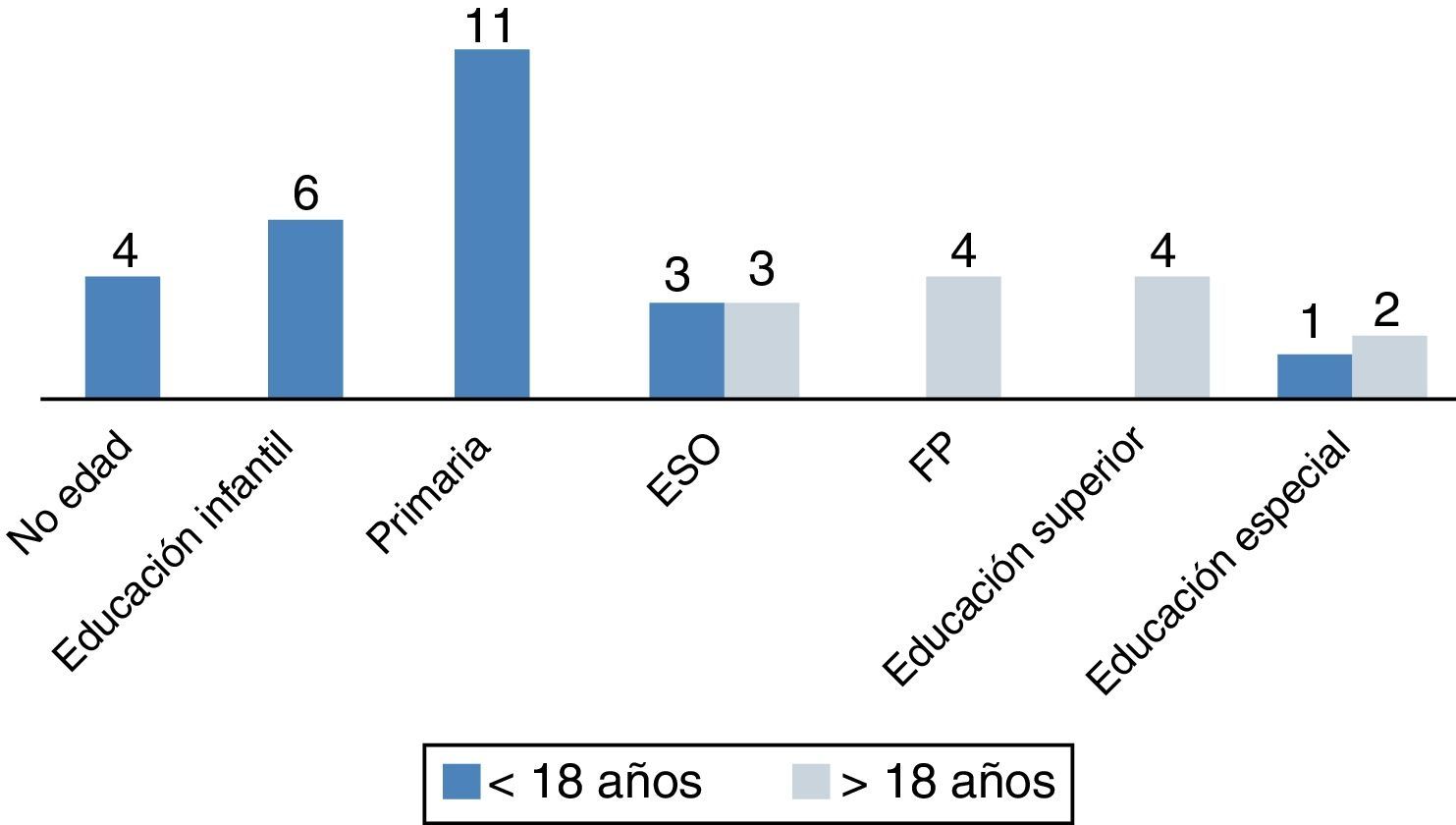
Este estudio agrupa por primera vez a los pacientes españoles con SHCC. La implementación del Registro europeo en España ha requerido el esfuerzo y la colaboración de 19 hospitales y 22 profesionales sanitarios para reclutar a 38 pacientes, al tratarse de una enfermedad muy rara y no existir aún centros de referencia nacionales para el seguimiento multidisciplinar, coordinado con los médicos regionales, como se recomienda2, y se lleva a cabo internacionalmente (Francia24, EE. UU.27). La puesta en marcha del laboratorio de referencia español para diagnóstico genético de SHCC es el primer paso dado en ese proyecto, que constituye un área de mejora.
Un tercio de los pacientes españoles son mayores de 18 años (todos diagnosticados en la infancia, muchos al nacer), lo que demuestra su larga supervivencia gracias a la VM domiciliaria y al cuidado continuado de sus familias. La entrada en la edad adulta plantea la difícil transición de estos niños crónicos desde los servicios de Pediatría (donde generalmente han recibido seguimiento estrecho) a Unidades de adultos, que desconocen esta patología, pudiendo quedar insuficientemente protegidos. Por ello, algunos países extienden la atención a pacientes mayores de edad en sus centros de referencia, en colaboración con especialistas de adultos27. La vida independiente y autónoma, las relaciones de pareja y la inserción en la vida laboral suponen un desafío para estos jóvenes que duermen con respirador.
El desarrollo neurocognitivo de los pacientes con SHCC está condicionado por aspectos relacionados con la propia enfermedad, por procesos intercurrentes y por el riesgo permanente de hipoxemia e hipercapnia, especialmente durante el sueño28-30. Aun así, la mayoría de los niños españoles han sido escolarizados en centros normales y 4 han alcanzado el nivel superior educativo, aunque un tercio han precisado refuerzo educativo mantenido. Por ello, la optimización de la atención de los niños españoles con SHCC implica su valoración psicopedagógica y curricular periódica, para detectar y tratar precozmente deficiencias de aprendizaje.
La frecuencia de mutaciones del presente estudio se corresponde con otras series publicadas y aparentemente también la correlación genotipo-fenotipo14, aunque, como se señala anteriormente, el escaso número de pacientes NPARM impide estudios comparativos; no obstante, en esta serie, los tumores del SNA están ausentes en pacientes PARM y la enfermedad de Hirschsprung predomina en pacientes 20/33 y NPARM. Por ello, el análisis genético es fundamental para diagnosticar la enfermedad y orientar en el tratamiento multidisciplinar y el seguimiento periódico protocolizado según genotipo, como recomienda la American Thoracic Society (ATS)2,15.
Respecto a otras patologías asociadas, las hipoglucemias aparecen en 5 pacientes (13%), frente al 8% de la serie de Vanderlaan et al.8; se trata de una complicación poco reseñada en la literatura, incluso no mencionada en publicaciones de expertos2; sin embargo, comporta gran relevancia clínica, porque las hipoglucemias perturban enormemente la vida diaria del niño y familia, tanto por los síntomas como por las medidas necesarias para su difícil control31. Dos pacientes (genotipos 20/27, 20/33) precisaron laparotomía urgente por presentar vólvulo gastrointestinal, complicación no descrita anteriormente por otros autores, aunque pudiera haberse englobado en las alteraciones de motilidad intestinal. Las patologías oculares, sin precisar tipo, son muy frecuentes, presentándose en casi la mitad de los pacientes españoles, al igual que en la serie de Vaderlaan et al.8. Patwari et al.5 describen alteración de la capacidad de adaptación pupilar a la luz. Por tanto, la exploración oftalmológica periódica incluyendo pupilometría es necesaria para prevenir la repercusión negativa de la alteración visual sobre el aprendizaje y desarrollo.
Los síntomas de hipoventilación central pueden pasar desapercibidos o ser malinterpretados y diagnosticarse tardíamente, sobre todo en mutaciones menos graves, como ocurrió en 3 de nuestros pacientes con genotipo 20/25, aunque también sucedió en un lactante 20/33, diagnosticado inicialmente de enfermedad de Hirschsprung. El retraso diagnóstico implica hipoxia e hipercapnia crónicas, que entorpecen el neurodesarrollo20; su prevención representa un área de mejora que conlleva mantener alto índice de sospecha ante hallazgos clínicos no aclarados, tales como episodios de cianosis, letargia, hipertensión pulmonar, necesidad de VM por infecciones leves o tras anestesia, convulsiones o retraso del neurodesarrollo.
La elección del modo ventilatorio en cada paciente y momento supone un aspecto muy relevante en la optimización del tratamiento. La TQ es la técnica más usada en los niños españoles con SHCC en la primera década de la vida, como en otras series8. Diversos autores y la ATS recomiendan la TQ en los primeros años de la vida, por seguridad y para garantizar un óptimo neurodesarrollo, evitando el cambio a VNI antes de los 6-8 años15. Sin embargo, es un asunto controvertido porque no hay estudios que analicen la influencia de la técnica ventilatoria sobre el neurodesarrollo. Costa Orvay et al.32 describen el fracaso de la VNI en 2 lactantes con SHCC frente a la experiencia positiva de otros autores33; en la serie de Vanderlaan et al., de 196 pacientes, el 14,3% nunca usó TQ8, porcentaje similar al obtenido en el presente estudio (15,8%). Este muestra a 8 pacientes tratados desde el inicio y dados de alta a domicilio con ventilación con mascarilla, sin TQ, 5 de los cuales habían presentado comienzo neonatal: los 2 con genotipo más grave (20/33, NPARM) precisaron transferir a TQ por presentar parada cardiorrespiratoria en su domicilio, frente a los 3 restantes, con mutaciones más leves (20/25, 20/25, no mutación), que han mantenido la mascarilla. Cuando se usa VNI en niños pequeños, además de la gravedad del genotipo, otro aspecto a considerar es el efecto negativo a largo plazo de la interfase sobre el crecimiento del macizo facial34,35, hecho no analizado en nuestro estudio. Por todo lo anterior, parece prudente utilizar la TQ como opción inicial en niños con SHCC de comienzo precoz, planteando la VNI como opción inicial solo en pacientes muy seleccionados, como pueden ser algunos bebés con genotipo menos grave determinante de menor hipoventilación y no comorbilidad, o en pacientes con comienzo tardío. El uso de la VNI siempre debe acompañarse de vigilancia estrecha y de medidas preventivas dirigidas a evitar deformidades faciales (alternancia de interfases, uso de mascarilla facial total o de presión negativa)36,37.
El MF asociado a otros modos es utilizado por solo 3 pacientes españoles, todos dependientes de VM 24 h, como otros autores refieren38. No obstante, el MF aislado se usa por adolescentes o adultos ventilados solo durante el sueño8,27, liberados de TQ y mascarilla. Los inconvenientes del MF están en relación con su implantación quirúrgica y con complicaciones técnicas o infecciosas39. Un nuevo sistema de marcapasos que estimula directamente el diafragma podría mejorar el procedimiento, aunque la experiencia aún es muy escasa40.
El paso de TQ a VNI se llevó a cabo en nuestros pacientes a la edad media de 13,7 años, el doble de la edad mínima de seguridad recomendada (6-8 años)15; solo un niño cambió antes de los 10 años; diversos factores no analizados en este estudio influyen en la decisión personalizada del momento de cambio: ventajas/inconvenientes, disposición y deseo del paciente o padres, experiencia del equipo médico.
La principal limitación del presente estudio es que el Registro no ha reclutado a todos los pacientes españoles con SHCC, porque algunas familias han rechazado participar y porque probablemente existen pacientes no diagnosticados o desconocidos. Con los pacientes conocidos y los datos demográficos publicados por el Instituto Nacional de Estadística (http://www.ine.es/inebmenu/mnu_dinamicapob.htm), la incidencia estimada en España del SHCC sería de 0,35/100.000 RN vivos; sin embargo, tomando como referencia la incidencia francesa24, en España nacerían cada año unos 2 pacientes con SHCC y sumarían unos 17 los aún no identificados. Sería necesario aumentar los esfuerzos colaborativos para identificarlos.
En resumen, la puesta en funcionamiento del Registro europeo de pacientes con SHCC en España ha permitido reconocer aspectos relevantes para optimizar sus cuidados: ausencia de centros de referencia en España, importancia del estudio genético y del refuerzo educativo y algunas limitaciones de los modos ventilatorios.
FinanciaciónEstudio financiado parcialmente por Proyecto FIS08/90233 (Angel Campos Barros). El diseño, elaboración y mantenimiento del Registro europeo fueron financiados por la Agencia Ejecutiva de Sanidad y Consumo de la Unión Europea.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los pacientes con SHCC y a sus familias. Al Consorcio Europeo del Síndrome de Hipoventilación Central.
