







Analizar las variaciones en el tratamiento del meduloblastoma, el tumor cerebral más frecuente en la infancia, y su repercusión en la supervivencia durante las 2 últimas décadas, así como sus características clínicas y anatomopatológicas.
Pacientes y métodosAnálisis de supervivencia de todos los casos de meduloblastoma diagnosticados en menores de 14 años desde enero de 1990 hasta diciembre del 2013 en una Unidad de Oncología Pediátrica.
ResultadosSesenta y tres pacientes fueron diagnosticados y tratados de meduloblastoma. La mediana de seguimiento fue 5,1 años (rango 0,65-21,7 años). La supervivencia global (SG) a los 3 y 5 años fue 66±13% y 55±14%, respectivamente. En los pacientes diagnosticados en la década de los 90, la SG a los 5 años fue 44%±25%, observándose un ascenso hasta 70±23% (p = 0,032) a partir del año 2000. En el modelo de regresión logística se incluyeron los factores clínicos implicados en el pronóstico: edad (p = 0,008), presencia de metástasis y/o resto tumoral (p = 0,007) y haber recibido quimioterapia, junto a radioterapia, tras la cirugía (p = 0,008), observándose diferencias estadísticamente significativas para todos ellos.
ConclusiónEn la última década se ha producido un importante aumento de la supervivencia del meduloblastoma en nuestro centro. En el análisis multivariante se observó que esta mejoría no estaba relacionada con la fecha de diagnóstico, sino con la introducción de la quimioterapia en el tratamiento adyuvante. Se confirmó que los factores clínicos relacionados significativamente con un peor pronóstico son la edad y la presencia de metástasis al diagnóstico.
The aim of the study is to analyse variations in the treatment of medulloblastoma, the most common childhood brain tumour, and its impact on survival over the past two decades, as well as its clinical and pathological features.
Patients and methodsSurvival analysis of all patients under 14 years old diagnosed with medulloblastoma between January 1990 and December 2013 in a Paediatric Oncology Unit.
ResultsSixty-three patients were diagnosed and treated for medulloblastoma, with a median follow-up of 5.1 years (range 0.65-21.7 years). The overall survival (OS) at 3 and 5 years was 66±13% and 55±14%, respectively.
The OS at 5 years was 44%±25% in patients diagnosed in the 1990's, showing an increase to 70%±23% (p=0.032) since 2000. Clinical prognosis factors were included in the logistic regression model: age (p=0.008), presence of metastases and/or residual tumour (p=0.007), and receiving chemotherapy with radiotherapy after surgery (p=0.008). Statistically significant differences were observed for all of them.
ConclusionIn our institution there has been a significant increase in medulloblastoma survival in the last decades. Multivariate analysis showed that this improvement was not related to the date of diagnosis, but with the introduction of chemotherapy in adjuvant treatment. This study confirmed that clinical factors significantly associated with worse outcome were age and presence of metastases at diagnosis.
Los tumores del sistema nervioso central (SNC) son los tumores sólidos más frecuentes en niños y la primera causa de muerte relacionada con cáncer en la infancia1.
Los tumores de origen embrionario suponen aproximadamente el 25% de todos los tumores primarios del SNC en menores de 18 años2. Según la WHO, podemos clasificarlos en 3 grupos: meduloblastoma, tumores neuroectodérmicos primitivos supratentoriales (sPNET)3.
El meduloblastoma es el tumor cerebral más frecuente en los niños, supone el 15-20% de todos los tumores del SNC. Es de origen neuroectodérmico y de localización infratentorial. Aproximadamente el 75% asienta en el vermis cerebeloso y crece hacia el cuarto ventrículo. Es más frecuente en los varones (≈ 65%) y la mediana de edad de presentación se sitúa alrededor de los 7 años3. Clásicamente, el meduloblastoma se ha clasificado según su histología: clásico, desmoplásico/nodular, de extensa modularidad, anaplásico o de células grandes, observándose peor pronóstico en la variante anaplásica y de células grandes4, y pronóstico favorable en los meduloblastomas desmoplásicos.
Los síntomas derivados de la hipertensión intracraneal son la forma de presentación clínica más frecuente: cefalea, náuseas y vómitos, irritabilidad, etc., de semanas, e incluso meses, de evolución, aunque también pueden aparecer síntomas relacionados con la localización tumoral: alteraciones del equilibrio, ataxia, dismetría, parálisis del vi par, etc.5.
El diagnóstico inicial se realiza mediante tomografía computarizada (TC) y/o resonancia magnética (RM). La TC permite evaluar de forma urgente el tumor y sus complicaciones, pero el estudio de elección es la RM6. Hasta un 30% de los casos pueden estar diseminados a través el espacio subaracnoideo en el momento del diagnóstico, por lo que el estudio de extensión se completa con una RM espinal y con el análisis citológico del líquido cefalorraquídeo (LCR)7. Las metástasis extraaxiales son muy infrecuentes8.
El diagnóstico diferencial debe hacerse con otros tumores de la fosa posterior, como astrocitomas pilocíticos, ependimomas o AT/RT6,9. Por ello, el diagnóstico definitivo requiere confirmación histopatológica.
El pronóstico de este tumor está estrechamente relacionado con 3 factores clínicos principales: la edad del paciente, la presencia de metástasis en el momento del diagnóstico y la existencia de tumor residual tras la cirugía, observándose peor pronóstico en menores de 3-5 años, en pacientes con enfermedad diseminada o con resto tumoral > 1,5cm210,11.
Los avances en investigación de los últimos años han llevado a una reciente categorización molecular del meduloblastoma en 4 subgrupos: grupo Wnt/βcatenina, grupo Sonic-Hedgehog, grupo 3 y grupo 4, cada uno de ellos con características biológicas y clínicas diferentes12,13. Por ejemplo, el subgrupo 3 es el de peor pronóstico y se asocia con frecuencia a la amplificación myc12,14,15.
Los pilares del tratamiento son la cirugía, la radioterapia y la quimioterapia, con diferentes esquemas terapéuticos según los factores de riesgo mencionados. La exéresis tumoral, tan radical como sea posible, es la primera actitud terapéutica. Seguida en todos los casos, salvo en niños pequeños, de radioterapia craneoespinal con boost en la fosa posterior a mayor o menor dosis, según el riesgo. La quimioterapia (derivados del platino, lomustina, metotrexato, etc.) ha demostrado su utilidad como tratamiento adyuvante desde los años 90, su intensidad es determinante en los casos de alto riesgo o en aquellos pacientes que no pueden recibir radioterapia por sus importantes secuelas, como los menores de 5 años.
En esta revisión vamos a analizar los factores clínicos implicados en el pronóstico del meduloblastoma, las variaciones en su tratamiento a lo largo de las últimas décadas y su repercusión en la supervivencia.
Pacientes y métodoRevisión retrospectiva de todos los pacientes ≤ 14 años con diagnóstico histopatológico de meduloblastoma tratados en la Unidad de Oncología Pediátrica del Hospital La Fe entre enero de 1990 y diciembre del 2013. Todos los casos fueron confirmados histológicamente por un patólogo especialista en tumores pediátricos. Se excluyó a los pacientes remitidos desde otros centros hospitalarios para trasplante de progenitores hematopoyéticos o radioterapia. El estudio de extensión se realizó mediante RM craneoespinal (excepto 1 pacientes de 1990 en los que se realizó TC craneal) y análisis citológico del LCR. La extensión de la enfermedad se estableció según la clasificación de Chang16 entendiéndose como enfermedad metastásica cualquier meduloblastoma con citología positiva para células tumorales y/o con diseminación en el espacio subaracnoideo supra/infratentorial diagnosticada por pruebas de imagen. Todos los pacientes fueron tratados según protocolos consecutivos de la International Society of Pediatric Oncology (SIOP) en función de los distintos factores de riesgo. Durante los 90, los protocolos de tratamiento del meduloblastoma de riesgo estándar evaluaban, de forma aleatorizada, la eficacia de añadir quimioterapia adyuvante17-20. Por ello, tras la cirugía, algunos pacientes recibieron exclusivamente radioterapia y otros quimioterapia y radioterapia. Los niños menores de 3-5 años fueron tratados únicamente con quimioterapia tras la exéresis quirúrgica. Los meduloblastomas de alto riesgo se trataron con cirugía, radioterapia y quimioterapia a altas dosis19,21 (tabla 1). Para esta revisión se consideró de alto riesgo a aquellos pacientes con meduloblastoma metastásico y/o resto tumoral > 1,5cm2 y de riesgo estándar si la resección fue total o con resto < 1,5cm2 (subtotal) sin diseminación.
Protocolos vigentes durante los periodos revisados (1990-2013)
| Protocolo | Esquema de tratamiento |
|---|---|
| SIOP 1 | Cirugía+RT+/–QT (lomustina, vincristina) |
| SIOP PNET 3 | MB RE: cirugía+/–QT (vincristina, carboplatino, etopósido, ciclofosfamida)+RT MB AR: cirugía+QT (vincristina, carboplatino, etopósido, ciclofosfamida)+RT |
| HIT-SIOP PNET 4 MB RE | Cirugía+RT+8 ciclos QT (cisplatino, lomustina, vincristina) |
| Esquema HART MB AR | QT altas dosis (metotrexato, vincristina, etopósido, ciclofosfamida, carboplatino)+RT hiperfraccionada y acelerada a altas dosis (HART)+doble megaterapia (tiotepa) con trasplante de progenitores hematopoyéticos autólogo |
| Sehop<3años | Cirugía+QT (cisplatino, etopósido, vincristina, ciclofosfamida, metotrexato, carboplatino) |
| Esquema Head Start<3años | QT 3 ciclos (vincristina, etopósido, ciclofosfamida, cisplatino+/–metotrexato)+3 ciclos megaterapia (tiotepa, carboplatino) con trasplante autólogo |
MB AR: meduloblastoma alto riesgo; MB RE: meduloblastoma riesgo estándar; QT: quimioterapia; RT: radioterapia.
Se recogieron las siguientes variables: fecha de nacimiento/diagnóstico/recaída/defunción/último seguimiento, sexo, clínica al diagnóstico, localización radiológica, grado de extensión de la enfermedad (RM/LCR), subtipo histológico, esquema de tratamiento utilizado, presencia de enfermedad residual tras la cirugía, grado de respuesta al tratamiento y evolución.
Las variables categóricas se describieron con el recuento numérico (porcentaje) de cada categoría. Las variables continuas como media±desviación estándar (DE) si la distribución fue normal (p>0,05, prueba de Kolmogorov-Smirnov) o como mediana y rango si la distribución no fue normal. La estimación de la curva de supervivencia se realizó mediante el método de Kaplan-Meier, la comparación univariante mediante el test Log-Rank y la multivariante mediante regresión de Cox. En el análisis multivariante se incluyeron los factores pronóstico que mostraron diferencias significativas en el análisis univariante y aquellos factores clínicos tradicionalmente considerados de mal pronóstico. Se aceptó como límite de significación estadística una p ≤ 0,05. El análisis de los datos se realizó con el paquete estadístico SPSS 20.0 y las gráficas con R 3.0.2.
ResultadosEn el periodo analizado se diagnosticaron 86 tumores embrionarios del SNC en pacientes menores de 14 años. El 74,4% de estos tumores fueron meduloblastomas (n=63), el 24,4% sPnet (n=22), así como un único AT/RT (1,1%).
PacientesLa mediana de edad en el momento del diagnóstico fue 7 años (rango: 7 meses-14 años). Cuarenta y dos pacientes eran varones (66,7%) y 21 niñas (33,3%). La clínica más frecuente de presentación fueron los síntomas de hipertensión intracraneal: 50 pacientes presentaban vómitos (79,4%) y 45 cefalea (71,4%) en el momento del diagnóstico. Otros síntomas neurológicos relacionados con la localización tumoral fueron: alteraciones de la marcha o ataxia (36,5%), estrabismo o diplopía por afectación del vi par (12,7%) o clínica de afectación de otros pares craneales (25,4%): parálisis facial, hipoacusia, etc. Síntomas menos frecuentes fueron: convulsiones, somnolencia, irritabilidad, regresión psicomotriz, etc. Entre ellos cabe destacar a 2 pacientes que inicialmente presentaron torpeza motora; otro con disfagia y un paciente con problemas de aprendizaje de aparición súbita. Diecisiete pacientes (27%) precisaron colocación de derivación ventricular externa o válvula de derivación ventrículo-peritoneal previa a la exéresis quirúrgica por hidrocefalia secundaria al tumor. Las características de los pacientes se resumen en la tabla 2.
Características de los pacientes (n=63)
| Características | n (%) |
|---|---|
| Sexo | |
| Varón | 42 (66,7) |
| Mujer | 21 (33,3) |
| Síntomas al diagnóstico | |
| Vómitos | 50 (79,4) |
| Cefalea | 45 (71,4) |
| Ataxia/alteraciones de la marcha | 23 (36,5) |
| Hidrocefalia+sistema derivación | 17 (26,9) |
| Afectación de pares craneales | 14 (22,2) |
| Parálisis del vi par | 8 (12,7) |
| Otros | 16 (25,3) |
| Localización | |
| Vermis | 47 (74,6) |
| Hemisferio cerebeloso izquierdo | 15 (23,8) |
| Hemisferio cerebeloso derecho | 10 (15,8) |
| IV ventrículo | 36 (57,1) |
| Otros (pedúnculos cerebelosos…) | 6 (9,5) |
| Estadificación | |
| Localizado | 40 (63,4) |
| M1 (localizado, LCR +) | 3 (4,8) |
| Metastásico (M2-M4) | 20 (31,7) |
| Histología | |
| Mb clásico | 46 (71,4) |
| Mb desmoplásico/nodular | 15 (25,4) |
| Mb anaplásico | 1 (1,6) |
| Mb de células grandes | 1 (1,6) |
| Protocolo de tratamiento | |
| SIOP 1 | 7 (11,1) |
| SIOP 3 | 19 (30,2) |
| SIOP PNET 4 | 16 (25,3) |
| SEHOP<3 años | 7 (11,1) |
| Esquema HEAD START<3 años | 3 (4,8) |
| Esquema HART MILAN HR-MB | 9 (14,3) |
| Otros | 2 (3,2) |
Localización tumoral: la localización más frecuente del tumor fue el vermis cerebeloso (75%), seguido de los hemisferios cerebelosos (24% izquierdo, 16% derecho) y un 9,5% en otras estructuras cerebelosas, como los pedúnculos. El 57% de los meduloblastomas ocupaban el iv ventrículo. Veintiún pacientes (32%) presentaban diseminación radiológica en el momento del diagnóstico.
Cirugía: en 50 pacientes (82,5%) se consiguió una resección total/subtotal con una única cirugía. Se realizó un segundo acto quirúrgico en 7 pacientes, alcanzando 3 exéresis completas más.
Subtipo histológico: el subtipo más frecuente fue el meduloblastoma clásico (71%), seguido del meduloblastoma desmoplásico (25,4%) y, por último, 2 únicos casos de meduloblastoma anaplásico y de células grandes.
Estadificación: 24 pacientes fueron considerados de alto riesgo por la presencia de metástasis en las pruebas de imagen en el momento del diagnóstico (n=21, celularidad tumoral en LCR (n=7) y/o resto tumoral (n=10). Los 39 pacientes restantes fueron considerados de riesgo estándar.
Tratamiento adyuvante: 53 pacientes (84,2%) recibieron radioterapia posquirúrgica, 12 de ellos como único tratamiento adyuvante. Los 41 pacientes restantes recibieron radioterapia y quimioterapia según diferentes esquemas terapéuticos (tabla 1), de los cuales 13 fueron sometidos a megaterapia con rescate de progenitores hematopoyéticos autólogos en al menos una ocasión. Diez pacientes, todos ellos menores de 4 años, fueron tratados exclusivamente con quimioterapia tras la cirugía.
El 87,3% de los pacientes completó el tratamiento. En 6 casos (9,5%) se suspendió por progresión/recaída de la enfermedad. El resto (3,2%) abandonó el tratamiento por otras causas (decisión familiar, cambio de centro, etc.).
Análisis de supervivencia: en el momento actual, 35 pacientes se encuentran fuera de tratamiento con una mediana de seguimiento de 5,1 años (rango: 0,65-21,7 años). La supervivencia global (SG) a los 3 y 5 años es de 66±13% y 55±14% (fig. 1). La supervivencia libre de eventos es 48±13% y 43±14%, respectivamente.
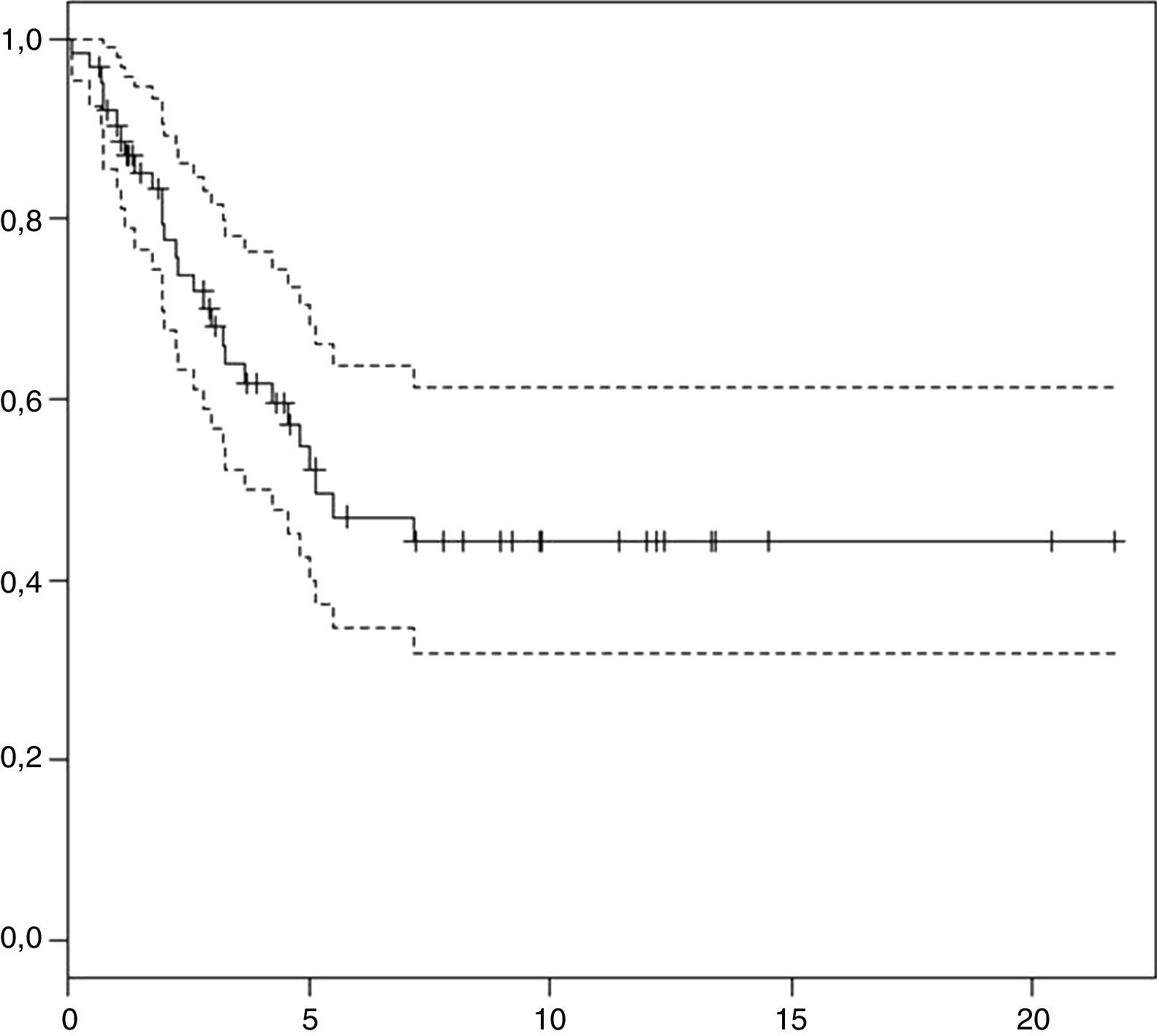
Durante el seguimiento, la mitad de los pacientes (n=32) recayeron o progresaron. De ellos, 7 se encuentran vivos y fuera de tratamiento. Todas las recaídas o progresiones de la enfermedad se produjeron durante los primeros 5 años de seguimiento, a excepción de un paciente que recayó de forma local a los 7 años y falleció tras varias líneas de tratamiento.
Cuatro pacientes (6,3%) fallecieron por toxicidad, todos ellos antes del año 2000. Dos durante la primera línea de tratamiento por complicaciones secundarias al trasplante y otros 2 pacientes durante el tratamiento de la recaída, uno por complicaciones del trasplante y otro por sepsis polimicrobiana e infección fúngica invasora.
En el análisis univariante por grupos de riesgo se observó una SG a los 5 años de 48±22% para el meduloblastoma de alto riesgo y de 61±17% para el grupo de riesgo estándar (p=0,11). En el análisis por factores de riesgo, el estadio en el momento del diagnóstico (p=0,018) y el tratamiento adyuvante con quimioterapia (p = 0,039) se correlacionan significativamente con la supervivencia. Destaca el aumento de la SG a los 5 años, que pasa del 44% en el periodo 1990-2000 al 70% entre 2000 y 2013 (p=0,032) (fig. 2).
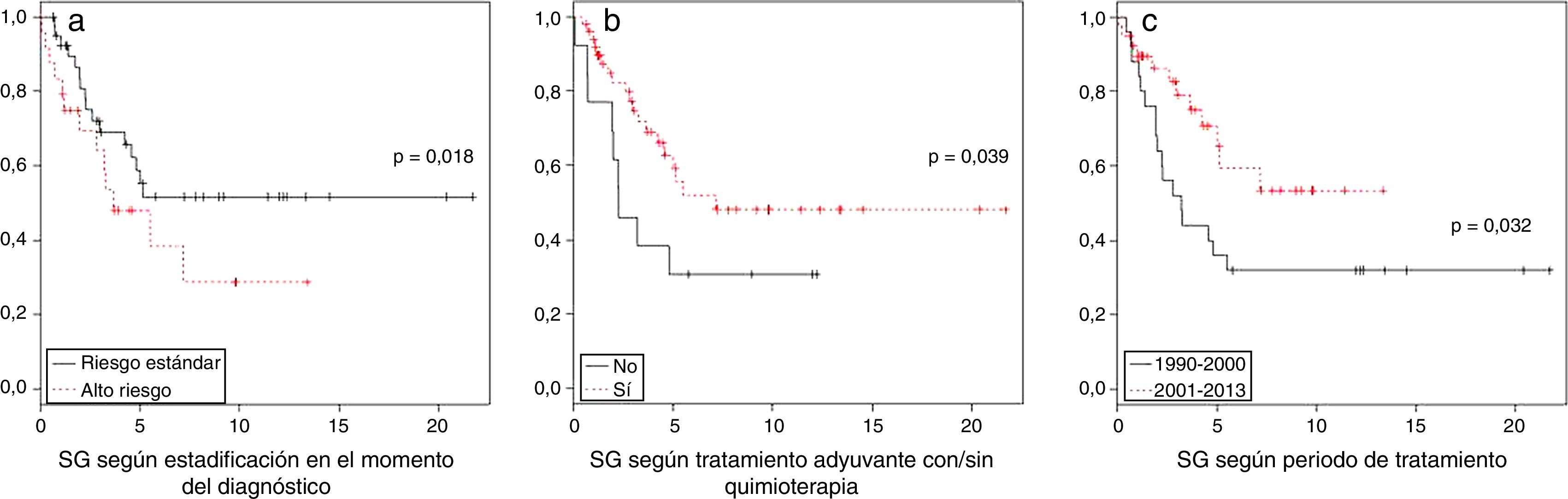
No se encontraron diferencias significativas según el grado de resección quirúrgica (p=0,348) ni entre los distintos subtipos histológicos (p=0,923).
En el análisis multivariante la edad (p=0,008) junto con la presencia de metástasis en el momento del diagnóstico y/o resto tumoral > 1,5cm2 (p=0,007) y la quimioterapia adyuvante (p=0,008) se correlacionan significativamente con la supervivencia, no así el periodo de tratamiento (p=0,467) (tabla 3).
DiscusiónEl meduloblastoma es el tumor cerebral más frecuente en la infancia. Su supervivencia ha mejorado en las últimas décadas gracias a la incorporación de la quimioterapia al tratamiento estándar con cirugía y radioterapia19,20,22, gracias a la intensificación del tratamiento del meduloblastoma de alto riesgo21,23-25 y gracias a la mejora de los tratamientos de soporte.
Este estudio analiza la experiencia de nuestro centro en el diagnóstico y manejo del meduloblastoma en pacientes pediátricos. Para ello, se han revisado retrospectivamente todos los casos de meduloblastoma diagnosticados y tratados en nuestra unidad desde 1990.
Las características de los pacientes (edad en el momento del diagnóstico, sexo, clínica, localización y presencia de metástasis) son similares a las descritas en la literatura3,5. La mediana de edad en el momento del diagnóstico es 7 años, con mayor prevalencia en varones. La clínica de presentación más frecuente son los síntomas derivados de la hipertensión intracraneal generada por el tumor. Generalmente, estos se acompañan de otros síntomas/signos neurológicos, pero no es raro que solamente aparezcan náuseas/vómitos o cefalea, ya que exclusivamente aparece a continuación de forma muy seguida. Diecisiete de nuestros pacientes presentaban exclusivamente vómitos y cefalea, 5 únicamente cefalea y otros 5 solo vómitos. Por tanto, ante una clínica inespecífica de vómitos o cefalea, especialmente si se asocian entre sí, debemos contemplar la posibilidad de un tumor intracraneal. La localización tumoral más frecuente es el vermis cerebeloso. Un tercio de los pacientes presentaban diseminación en el momento del diagnóstico y el número de casos en los que se alcanzó resección total/subtotal en nuestra serie es igual al publicado en la literatura20. El subgrupo histológico más común, tal y como recogen otras series, es el meduloblastoma clásico, seguido por el desmoplásico4,26.
La SG del meduloblastoma en nuestra serie a los 3 años es 66±13%, lo que coincide con la SG de este tumor en España según datos del Registro Nacional de Tumores Infantiles (65±17%)27.
Respecto al grupo clasificado como riesgo estándar, la supervivencia a los 3 años es del 68±16%, ligeramente inferior al 70-80% descrito por otros grupos internacionales, posiblemente debido al tamaño muestral y al amplio periodo analizado. Esto último supone variaciones en las clasificaciones de riesgo y en la estrategia terapéutica, lo que en muestras pequeñas lleva a pequeñas desviaciones respecto al resultado de otros grupos17-20,22,28.
En los pacientes de alto riesgo, la SG a los 3 años es del 64±20%, similar a algunos datos publicados donde la supervivencia para este grupo no supera el 60-70%23,24.
En el análisis univariante no se observan diferencias estadísticamente significativas entre ambos grupos de riesgo dado que existen factores de confusión como la edad y las variaciones en el tratamiento adyuvante que deben ser tenidos en cuenta.
Al analizar los factores clínicos de riesgo por separado, se confirma un peor pronóstico en los pacientes con diseminación en el momento del diagnóstico (p=0,017). La supervivencia es mayor en los pacientes con exéresis tumoral completa respecto a aquellos con resto > 1,5cm2, pero las diferencias no son estadísticamente significativas (p=0,348), probablemente debido al escaso número de pacientes con resto tumoral. No se han observado diferencias estadísticamente significativas entre los distintos grupos histológicos, dado que únicamente 2 pacientes fueron diagnosticados de meduloblastoma anaplásico/de células grandes este dato no es valorable.
Durante el periodo revisado se observa una clara mejoría de la supervivencia en la última década, pasando de una SG a los 5 años del 44±25% al 70±23% en los pacientes diagnosticados a partir del año 2000 (p 0,032), similar a la de otras series publicadas20,22. Esta mejoría en la supervivencia se debe principalmente al uso sistemático de la quimioterapia en el tratamiento estándar del meduloblastoma (p=0,000), no habiendo diferencias significativas en el resto de factores clínicos de riesgo entre ambos periodos (edad, resto tumoral y diseminación en el momento del diagnóstico).
En el análisis multivariante la mejora de supervivencia entre ambas décadas pierde su significación estadística al incluirse en el modelo la quimioterapia. Sin embargo, sí se observan diferencias estadísticamente significativas para todos los demás factores clínicos de riesgo: la edad y la presencia de resto y/o diseminación en el momento del diagnóstico, que influyen negativamente en la supervivencia (tabla 3).
Por tanto, podemos concluir que los factores clave en la mejoría de la supervivencia en las últimas décadas son: la incorporación de la quimioterapia a los esquemas terapéuticos, algo claramente demostrado por todos los grupos cooperativos (SIOP, COG) en la década de los 90, y el aumento de la intensidad de los mismos. No obstante, no puede desestimarse la influencia de los avances en el tratamiento radioterápico y en las técnicas de imagen, ya que en la actualidad puede realizarse una estadificación más precisa y una mejor evaluación de la resección tumoral.
En el futuro, los esfuerzos deben concentrarse en ajustar la intensidad del tratamiento, tanto a los factores clínicos de riesgo, como al riesgo biológico del tumor para minimizar sus efectos secundarios. Hasta hace pocos años la biología del meduloblastoma era poco conocida, sin embargo, cada vez se incorporan más estudios biológicos en el momento del diagnóstico de este tumor que permiten categorizarlo mejor29. Esta revisión no incluye los estudios biológicos, ya que se han incorporado recientemente a la práctica clínica habitual. Futuras publicaciones deberán tener en cuenta todos estos aspectos para poder analizar de forma integral el comportamiento del meduloblastoma.
FinanciaciónInstituto de Investigación Sanitaria La Fe. Asociación Pablo Ugarte.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
