




The aim of the study is to analyse variations in the treatment of medulloblastoma, the most common childhood brain tumour, and its impact on survival over the past two decades, as well as its clinical and pathological features.
Patients and methodsSurvival analysis of all patients under 14 years old diagnosed with medulloblastoma between January 1990 and December 2013 in a Paediatric Oncology Unit.
ResultsSixty-three patients were diagnosed and treated for medulloblastoma, with a median follow-up of 5.1 years (range 0.65–21.7 years). The overall survival (OS) at 3 and 5 years was 66±13% and 55±14%, respectively.
The OS at 5 years was 44±25% in patients diagnosed in the 1990s, showing an increase to 70±23% (P=.032) since 2000. Clinical prognosis factors were included in the logistic regression model: age (P=.008), presence of metastases and/or residual tumour (P=.007), and receiving chemotherapy with radiotherapy after surgery (P=.008). Statistically significant differences were observed for all of them.
ConclusionIn our institution there has been a significant increase in medulloblastoma survival in the last decades. Multivariate analysis showed that this improvement was not related to the date of diagnosis, but with the introduction of chemotherapy in adjuvant treatment. This study confirmed that clinical factors significantly associated with worse outcome were age and presence of metastases at diagnosis.
Analizar las variaciones en el tratamiento del meduloblastoma, el tumor cerebral más frecuente en la infancia, y su repercusión en la supervivencia durante las 2 últimas décadas, así como sus características clínicas y anatomopatológicas.
Pacientes y métodosAnálisis de supervivencia de todos los casos de meduloblastoma diagnosticados en menores de 14 años desde enero de 1990 hasta diciembre del 2013 en una Unidad de Oncología Pediátrica.
ResultadosSesenta y tres pacientes fueron diagnosticados y tratados de meduloblastoma. La mediana de seguimiento fue 5,1 años (rango 0,65-21,7 años). La supervivencia global (SG) a los 3 y 5 años fue 66±13% y 55±14%, respectivamente. En los pacientes diagnosticados en la década de los 90, la SG a los 5 años fue 44%±25%, observándose un ascenso hasta 70±23% (p = 0,032) a partir del año 2000. En el modelo de regresión logística se incluyeron los factores clínicos implicados en el pronóstico: edad (p = 0,008), presencia de metástasis y/o resto tumoral (p = 0,007) y haber recibido quimioterapia, junto a radioterapia, tras la cirugía (p = 0,008), observándose diferencias estadísticamente significativas para todos ellos.
ConclusiónEn la última década se ha producido un importante aumento de la supervivencia del meduloblastoma en nuestro centro. En el análisis multivariante se observó que esta mejoría no estaba relacionada con la fecha de diagnóstico, sino con la introducción de la quimioterapia en el tratamiento adyuvante. Se confirmó que los factores clínicos relacionados significativamente con un peor pronóstico son la edad y la presencia de metástasis al diagnóstico.
Tumours of the central nervous system (CNS) are the most frequent solid tumours and the leading cause of cancer-related death in children.1
Embryonal tumours account for approximately 25% of all primary CNS tumours in individuals aged less than 18 years.2 According to WHO classification, they can be categorized into three groups: medulloblastoma, CNS supratentorial primitive neuroectodermal tumor (sPNET) and atypical teratoid/rhabdoid tumour (AT/RT).3
Medulloblastoma is the most frequent brain tumour in children and accounts for 15–20% of all CNS tumours. It has a neuroectodermal origin and an infratentorial location. Approximately 75% arise in the cerebellar vermis and grow towards the fourth ventricle. It is more frequent in males (≈65%) and the median age of onset is approximately 7 years.3 Historically, medulloblastoma has been classified based on histology: classical, desmoplastic/nodular, with extensive nodularity, anaplastic or large cell, with the worst prognosis corresponding to the anaplastic and large cell category, and a favourable prognosis in desmoplastic medulloblastoma.
The most common clinical presentation consists of symptoms caused by raised intracranial pressure, such as headache, nausea and vomiting or irritability, lasting weeks or even months, although other symptoms may also manifest depending on tumour location, such as changes in balance, ataxia, dysmetria, sixth cranial nerve palsy, etc.5
The initial diagnosis is made by computed tomography (CT) and magnetic resonance imaging (MRI). Computed tomography allows for the urgent evaluation of the tumour and its complications, but MRI is the gold standard.6 Up to 30% of patients may have dissemination through the subarachnoid space at the time of diagnosis, so diagnostic staging is completed by MRI of the spine and histological assessment of cerebrospinal fluid (CSF).7 Extra-axial metastases are very rare.8
The differential diagnosis must include other tumours of the posterior fossa, such as pilocytic astrocytoma, ependymoma or AT/RT.6,9 Thus, the diagnosis must be confirmed by histological assessment.
The clinical prognostication of medulloblastoma is intimately associated to three key factors: patient age, the presence of metastasis at the time of diagnosis, and the degree of residual disease after surgery, with a poorer prognosis in patients aged less than 3–5 years, with disseminated disease, or with more than 1.5cm2 of residual tumour.10,11
Advances in research in recent years have led to a new classification scheme for medulloblastoma that divides it into four molecular subgroups: Wnt/βcatenin group, Sonic Hedgehog group, group 3 and group 4, each of which has different biological and clinical characteristics.12,13 For example, group 3 has the poorest prognosis and is frequently associated with MYC amplification.12,14,15
The cornerstones of treatment are surgery, radiotherapy and chemotherapy, with different treatment schemes based on the aforementioned risk factors. The first line of treatment is tumour resection, as extensive as possible. This is followed in all cases, save in very young children, by craniospinal irradiation with posterior fossa boost treatment at varying doses based on risk. Chemotherapy (platinum derivatives, lomustine, methotrexate, etc.) has proven useful as an adjuvant therapy since the 1990s, and its intensification is a decisive factor in high-risk cases or in patients that cannot undergo radiotherapy due to their significant sequelae, such as children aged less than 5 years.
In this review, we will analyse the clinical factors involved in the prognostication of medulloblastoma and the changes in the management of medulloblastoma in recent decades along with their impact on survival.
Patients and methodsWe conducted a retrospective review of all patients aged 14 years or less with a histopathological diagnosis of medulloblastoma treated in the Paediatric Oncology Unit of the Hospital La Fe between January 1990 and December 2013. A pathologist specialised in paediatric tumours confirmed all cases by histological assessment. We excluded patients referred by other hospitals for hematopoietic stem cell transplantation or radiotherapy. Staging was investigated by craniospinal MRI (except in one patient in 1990, in which staging was determined by cranial CT) and CSF cytology. The extent of disease was established using the Chang system,16 and we defined metastatic disease as any medulloblastoma with detection of tumour cells in CSF cytology and/or with dissemination in the infra- or supratentorial subarachnoid space diagnosed by means of imaging tests. All patients were treated based on their risk factors following consecutive protocols of the International Society of Paediatric Oncology (SIOP). In the 1990s, trial protocols for average-risk medulloblastoma evaluated the efficacy of adding adjuvant chemotherapy in randomly assigned patients.17–20 Thus, following surgery, some patients were treated exclusively with radiotherapy and others with a combination of chemotherapy and radiotherapy. Children aged less than 3–5 years were treated with chemotherapy alone following surgical resection. High-risk medulloblastomas were treated with surgery, radiotherapy and high-dose chemotherapy19,21 (Table 1). In this review, we defined high-risk patients as those with metastatic medulloblastoma and/or more than 1.5cm2 of residual tumour, and average-risk patients as those in whom total or near-total (residual tumour <1.5cm2) resection was achieved and without metastatic disease.
Protocols applied during the reviewed periods (1990–2013).
| Protocol | Treatment scheme |
|---|---|
| SIOP 1 | Surgery+RTX±CTX (lomustine, vincristine) |
| SIOP PNET 3 | AR MB: Surgery±CTX (vincristine, carboplatin, etoposide, cyclophosphamide)+RTX MB AR: Surgery+CTX (vincristine, carboplatin, etoposide, cyclophosphamide)+RTX |
| HIT-SIOP PNET 4 for AR MB | Surgery+RTX+8 cycles of CTX (cisplatin, lomustine, vincristine) |
| HART for HR MB | High-dose CTX (methotrexate, vincristine, etoposide, cyclophosphamide, carboplatin)+hyperfractionated and accelerated RTX (HART)+double megatherapy (thiotepa) autologous haematopoietic stem cell transplantation |
| SEHOP<3 years | Surgery+CTX (cisplatin, etoposide, vincristine, cyclophosphamide, methotrexate, carboplatin) |
| Head Start<3 years | 3 cycles of CTX (vincristine, etoposide, cyclophosphamide, cisplatin±methotrexate)+3 cycles megatherapy (thiotepa, carboplatin) with autologous transplant |
AR MB, average risk medulloblastoma; CTX, chemotherapy; HR MB, high-risk medulloblastoma; RTX,radiotherapy.
We collected data for the following variables: dates of birth, diagnosis, relapse, death and last followup; sex; clinical manifestations at diagnosis, radiologic location, extent of disease (MRI/CSL cytology), histological variant, treatment protocol used, presence of residual disease after surgery, degree of response to treatment, and outcome.
We have expressed categorical variables as absolute frequencies and percentages, and continuous variables as mean±standard deviation (SD) if they followed a normal distribution (P>.05 in the Kolmogorov–Smirnov test) or as median and range if the distribution was not normal. We estimated survival curves using the Kaplan–Meier method, performed univariate contrasts by means of the log-rank test, and used Cox regression for the multivariate analysis. The factors included in the multivariate analysis were those that had shown significant differences in the univariate analysis as well as clinical factors that have been traditionally considered as indicators of a poor prognosis. We defined statistical significance as P≤.05. We performed the statistical analysis with the statistical package SPSS version 20.0, and generated the graphs with R version 3.0.2.
ResultsIn the period under study, 86 CNS embryonal tumours were diagnosed in patients aged less than 14 years. Of all these tumours, 74.4% were medulloblastomas (n=63), 24.4% sPNETs (n=22), and one was an AT/RT (1.1%).
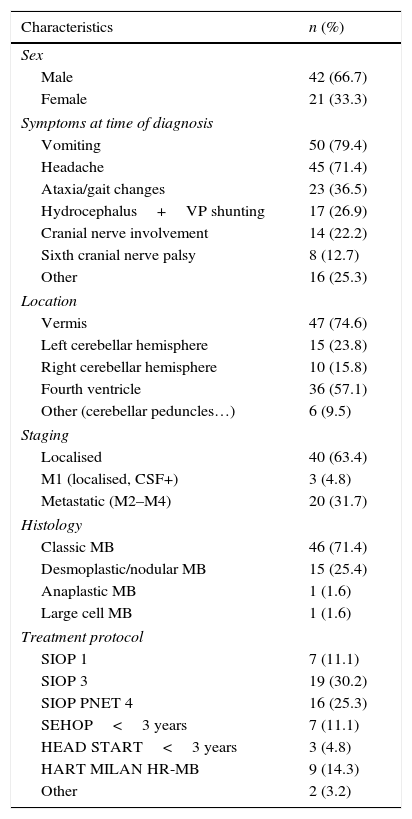
PatientsThe median age at diagnosis was 7 years (range, 7 months–14 years). Forty-two patients were male (66.7%) and 21 female (33.3%). The most frequent clinical presentation consisted of symptoms caused by raised intracranial pressure: 50 patients had vomiting (79.4%) and 45 had headache (71.4%) at the time of diagnosis. Other neurologic symptoms that were associated with tumour location included gait changes and ataxia (36.5%), ocular misalignment or diplopia due to sixth cranial nerve involvement (12.7%) or symptoms consistent with involvement of other cranial nerves (25.4%) such as facial paralysis, hearing loss, and others. Less frequent symptoms included convulsions, somnolence, irritability or psychomotor regression. Two of these patients presented with motor skill deficits; one with dysphagia, and one with learning impairments of abrupt onset. Seventeen patients (27%) required placement of a ventriculoperitoneal shunt prior to surgical resection for the management of hydrocephalus secondary to the tumour. Table 2 summarises the characteristics of the patients.
Patient characteristics (n=63).
| Characteristics | n (%) |
|---|---|
| Sex | |
| Male | 42 (66.7) |
| Female | 21 (33.3) |
| Symptoms at time of diagnosis | |
| Vomiting | 50 (79.4) |
| Headache | 45 (71.4) |
| Ataxia/gait changes | 23 (36.5) |
| Hydrocephalus+VP shunting | 17 (26.9) |
| Cranial nerve involvement | 14 (22.2) |
| Sixth cranial nerve palsy | 8 (12.7) |
| Other | 16 (25.3) |
| Location | |
| Vermis | 47 (74.6) |
| Left cerebellar hemisphere | 15 (23.8) |
| Right cerebellar hemisphere | 10 (15.8) |
| Fourth ventricle | 36 (57.1) |
| Other (cerebellar peduncles…) | 6 (9.5) |
| Staging | |
| Localised | 40 (63.4) |
| M1 (localised, CSF+) | 3 (4.8) |
| Metastatic (M2–M4) | 20 (31.7) |
| Histology | |
| Classic MB | 46 (71.4) |
| Desmoplastic/nodular MB | 15 (25.4) |
| Anaplastic MB | 1 (1.6) |
| Large cell MB | 1 (1.6) |
| Treatment protocol | |
| SIOP 1 | 7 (11.1) |
| SIOP 3 | 19 (30.2) |
| SIOP PNET 4 | 16 (25.3) |
| SEHOP<3 years | 7 (11.1) |
| HEAD START<3 years | 3 (4.8) |
| HART MILAN HR-MB | 9 (14.3) |
| Other | 2 (3.2) |
Tumour location: the most frequent tumour location was the cerebellar vermis (75%), followed by the cerebellar hemispheres (24% left, 16% right) and 9.5% in other cerebellar structures, such as the peduncles. Fifty-seven percent of the medulloblastomas occupied the fourth ventricle. Twenty-one patients (32%)had dissemination at the time of diagnosis, which was detected by radiology.
Surgery: total or near-total resection was achieved with a single surgery in 50 patients (82.5%). Seven patients underwent a second surgery, and total resection was achieved in three.
Histological subtypes: the most frequent subtype was classic medulloblastoma (71%), followed by desmoplastic medulloblastoma (25.4%) and, last of all, there were only two cases of anaplastic and large cell medulloblastoma.
Staging: 24 patients were considered high-risk due to the detection of disseminated disease by imaging tests at the time of diagnosis (n=21), presence of tumour cells in CSF (n=7) and/or presence of residual disease (n=10). The other 39 patients were classified as average-risk.
Adjuvant therapy: 53 patients (84.2%) received postsurgical radiotherapy, which was the sole adjuvant therapy in 12. The remaining 41 patients received radiotherapy and chemotherapy following different treatment protocols (Table 1), and 13 of them underwent megatherapy with autologous stem cell rescue at least once. Ten patients, all of them aged less than 4 years, were treated with chemotherapy alone following surgery.
Treatment was completed by 87.3% of patients. In six patients (9.5%), treatment was discontinued due to disease progression or recurrence. In the remaining patients (3.2%), treatment was discontinued for other reasons (choice of the family, change of medical facility, etc.).
Survival analysis: at present, 35 patients are not receiving treatment, with a median duration of followup of 5.1 years (range, 0.65–21.7 years). At 3 and 5 years from diagnosis, the overall survival (OS) was 66±13% and 55±14% (Fig. 1), and the event-free survival was 48±13% and 43±14%, respectively.
.")
During the followup, half of the patients (n=32) experienced a recurrence or disease progression. Of these patients, seven are still alive and are currently not receiving any treatment. Recurrences and disease progression happened in the first five years of followup in all patients, except in one patient that experienced a local recurrence at seven years and died after undergoing different lines of treatment.
Four patients (6.3%) died of toxicity, all of them before year 2000. Two died during initial treatment due to transplant-related complications, and another two during relapse treatment—one due to transplant complications and the other due to polymicrobial sepsis and invasive fungal infection.
The univariate analysis by risk groups found a 5-year OS of 48±22% in patients with high-risk medulloblastoma and 61±17% in patients with average-risk medulloblastoma (P=.11). In the analysis by risk factor, staging at the time of diagnosis (P=.018) and adjuvant chemotherapy (P=.039) were significantly associated with survival. We want to highlight the improvement in the 5-year overall survival, which increased from 44% in the 1990–2000 period to 70% between 2000 and 2013 (P=.032) (Fig. 2).

We did not find statistically significant differences based on the extent of surgical resection (P=.348) or histological subtype (P=.923).
In the multivariate analysis, patient age (P=.008) along with the presence of disseminated disease at the time of diagnosis and/or of more than 1.5cm2 of residual tumour (P=.007) and the use of adjuvant chemotherapy (P=.008) were significantly correlated to survival, while the period of treatment was not (P=.467) (Table 3).
DiscussionMedulloblastoma is the most frequent paediatric brain tumour. Its survival has improved in recent decades thanks to the addition of chemotherapy to the standard treatment with surgery and radiotherapy,19,20,22 intensification treatment in high-risk medulloblastoma,21,23–25 and advances in supportive care.
This study analyses the experience in our hospital in the diagnosis and management of medulloblastoma in paediatric patients. To do so, we did a retrospective review of all the cases of medulloblastoma diagnosed and treated in our unit since 1990.
Patient characteristics (age at the time of diagnosis, sex, clinical manifestations, tumour location and presence of metastases) were similar to those described in the literature.3,5 The median age at diagnosis was 7 years, and the prevalence was higher in males. The most frequent presenting symptoms are derived from the increased intracranial pressure caused by the tumour. Usually, these are accompanied by other neurologic symptoms and signs, but it is not rare for these patients to present exclusively nausea/vomiting or headache. Seventeen of our patients presented with vomiting and headache, five with only headache, and another five with only vomiting. Therefore, in patients with nonspecific presentations involving vomiting or headache, and especially combining both, we must contemplate the possibility of an intracranial tumour. The most frequent tumour location was the cerebellar vermis. One third of the patients had disseminated disease at the time of diagnosis, and the proportion of patients in whom total or near total resection was achieved in our series is the same as the proportion reported in the literature.20 The most frequent histological subtype, consistent with other published series, was classic medulloblastoma, followed by desmoplastic medulloblastoma.4,26
The 3-year OS of medulloblastoma patients in our series was 66±13%, which is consistent with the OS for this type of tumour in Spain based on data from the National Registry of Paediatric Tumours (65±17%).27
In patients classified as average-risk, the 3-year OS was 68±16%, somewhat lower than that reported by other international groups, which may be due to the small sample size and the long period under study. The latter involves changes in risk classifications and treatment approaches, which would lead to small deviations in small samples from the results obtained by other groups.17–20,22,28
In high-risk patients, the 3-year OS was 64±20%, consistent with other studies that reported a survival for this group of 60–70%.23,24
The univariate analysis did not find statistically significant differences between both risk groups, as there were confounding factors such as age and variability in adjuvant therapy that need to be taken into account.
When we analysed clinical risk factors separately, we confirmed that the prognosis was poorer in patients with disseminated disease at the time of diagnosis (P=.017). Survival was higher in patients with total tumour resection compared to those with more than 1.5cm2 of residual disease, but these differences were not statistically significant (P=.348), probably due to the small number of patients with residual tumours. We did not observe statistically significant differences between the different histological groups, as only two patients received a diagnosis of anaplastic/large cell medulloblastoma, which precluded the study of this factor.
In the period under study there was a clear improvement in survival in the last decade, with the 5-year OS increasing from 44±25% to 70±23% in patients that received their diagnosis starting at year 2000 (P=.032), similar to the survival reported in other case series.20,22 This improvement in survival is mostly due to the systematic use of chemotherapy in the standard treatment of medulloblastoma (P=.000), with no significant differences in survival between the two periods associated to any of the other clinical risk factors (age, residual disease and dissemination at the time of diagnosis).
The results of the multivariate analysis of changes in survival between these two decades stopped being statistically significant when chemotherapy was added to the model. However, we did find statistically significant differences for all other clinical risk factors: age, presence of residual disease and/or dissemination at the time of diagnosis, which had a negative impact on survival (Table 3).
Therefore, we can conclude that the key factors that have led to improved survival in recent decades were the incorporation of chemotherapy to treatment schemes, which has been clearly demonstrated by all collaborative research groups (SIOP, COG) in the 1990s, and the intensification of treatment. However, we cannot underestimate the influence of advances in radiotherapy and imaging techniques, as the stage of disease and the extent of tumour resection can be determined more accurately today.
Future efforts must focus on adjusting the intensity of treatment to clinical as well as biological risk factors to minimise adverse effects. Until a few years ago, the biology of medulloblastoma was poorly understood, but biological tests are increasingly included in the diagnosis of medulloblastoma that allow a better characterisation of the tumour.29 This review did not include biological tests, as it is only recently that they have been incorporated in everyday clinical practice. Future studies should take all of these aspects into account to make a comprehensive analysis of the behaviour of medulloblastoma.
FundingInstituto de Investigación Sanitaria La Fe. Asociación Pablo Ugarte.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Igual Estellés L, Berlanga Charriel P, Cañete Nieto A. Meduloblastoma: mejoría de la supervivencia en las últimas décadas. Experiencia de un centro. An Pediatr (Barc). 2017;86:4–10.
