




El angioma en penacho o tufted angioma (TA) es una tumoración vascular benigna poco frecuente, que suele aparecer en la infancia, aunque existen casos de aparición en la edad adulta. Su presentación clínica es muy variable. Se manifiesta típicamente como una mácula, pápula o nódulo eritematovioláceo en el tronco o el cuello. Histológicamente, se caracteriza por agregados de lóbulos angiomatosos en la dermis formando pequeños penachos de capilares.
Pacientes y métodosEstudio retrospectivo observacional de los casos diagnosticados de TA en los últimos 20 años en nuestro centro.
ResultadosPresentamos un total de 9 casos de angiomas en penacho en la infancia. El 77,7% de los casos fueron congénitos, lo que representa una frecuencia superior a la descrita previamente. Nuestros pacientes presentaron regresión espontánea en el 55,5% de los casos, siendo más frecuente en el grupo de TA congénitos. A diferencia de las otras series descritas en la literatura, observamos un mayor porcentaje de pacientes con regresión espontánea, un predominio femenino (6 de los 9 niños) y una localización más frecuente en miembros los superiores. Ninguno de nuestros pacientes presentó fenómeno de Kasabach-Merritt.
ConclusionesDada la alta tasa de involución espontánea en TA congénitos o tempranos, en ausencia de otras complicaciones, la vigilancia sería una buena opción de manejo, monitorizando estrechamente al paciente.
Tufted angioma (TA) is a rare benign vascular tumor that mostly appears during infancy or early childhood, although there are cases reported in adults. Clinical presentation and evolution of TA can vary. Histologically, it takes on a classic appearance of vascular tufts (“cannon ball” like appearance).
Patients and methodsA retrospective observational study was conducted that included all patients diagnosed with TA at our center in the last 20 years.
ResultsA series of 9 cases of tufted angioma in childhood are presented, 77.7% of which were congenital. This represents a frequency higher than previously described. Spontaneous regression was observed in 55.5% of the cases, and was more frequent in the congenital TA group. Unlike other TA series reported in the literature, a higher proportion of patients with spontaneous regression was observed in this series, with a higher prevalence in females (6 out of 9 children) and predominantly located in the upper limbs. None of our patients had Kasabach-Merritt phenomenon.
ConclusionsThere are many ways of treating TA, but none are uniformly effective. Given the high rate of spontaneous regression in congenital or early TA, we suggest that, in the absence of other complications, monitoring would be a good option for management.
El angioma en penacho o tufted angioma (TA) es una tumoración vascular benigna poco frecuente. Fue descrita por primera vez en 1949 por Nakagawa1 como angioblastoma. Posteriormente, Jones y Orkin1 en 1971 presentaron una serie de casos con características histopatológicas similares, acuñando el nombre de angioma en penacho. Este término deriva de su apariencia histopatológica en la que se observan pequeños «penachos» de capilares y lóbulos angiomatosos en la dermis asociados a vasos linfáticos dilatados, dando un patrón característico en perdigonada o «cannon ball»2. El TA puede ser congénito o adquirido, siendo la incidencia de casos congénitos mayor de lo que se pensaba inicialmente, llegando a ser el 50% en las últimas revisiones. El resto aparecen más frecuentemente durante los primeros años de vida, aunque hay casos descritos de aparición en la edad adulta.3,4 Clínicamente, suele tratarse de una lesión solitaria, aunque existen casos diseminados5 y se presenta como un nódulo o placa infiltrada eritematoviolácea, generalmente mal delimitada6. Suele ser una lesión asintomática, pudiendo producir molestias locales, calor, dolor o inflamación, siendo la complicación más grave el fenómeno de Kasabach-Merritt.
Pacientes y métodosRealizamos un estudio retrospectivo observacional y descriptivo en el que revisamos los casos de TA diagnosticados mediante biopsia en los últimos 20 años en el Hospital General Universitario de Valencia en menores de 15 años. Para la selección de pacientes se revisaron las historias clínicas en papel y se utilizó el sistema informático Intranet de registro de informes de nuestro hospital. Se excluyó a los pacientes con edad superior a 15 años al inicio de la enfermedad y a aquellos pacientes que no tenían un diagnóstico anatomopatológico.
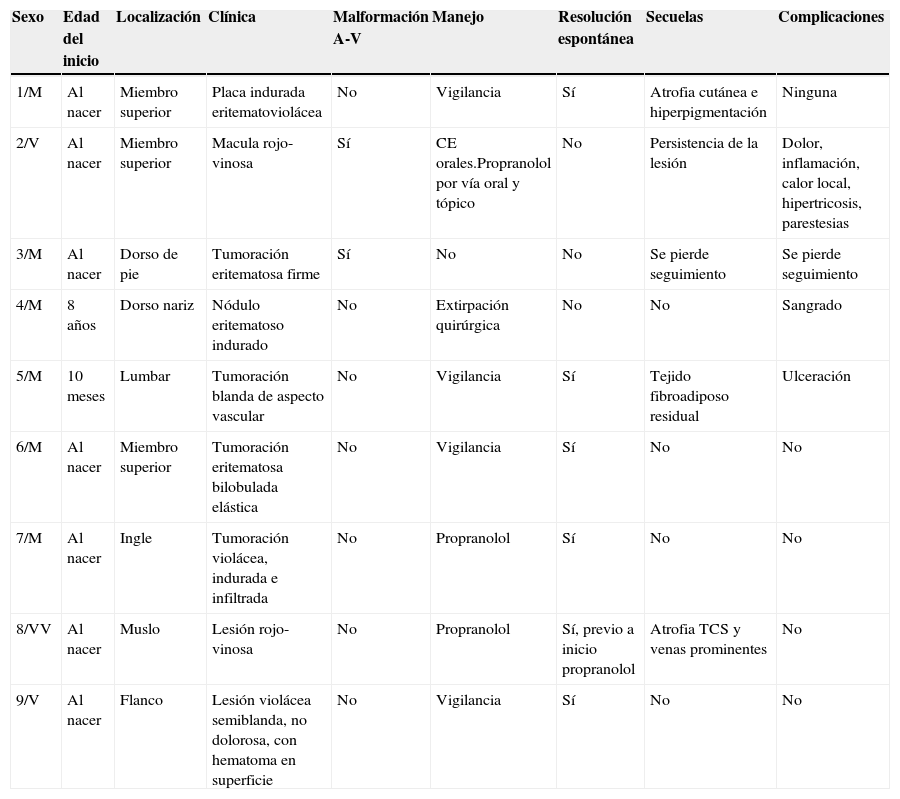
Se ha incluido a un total de 9 pacientes, de los cuales se han recogidos datos epidemiológicos, como sexo, edad de inicio y raza; datos de presentación clínica, como localización, apariencia, diagnóstico clínico; datos de metodología diagnóstica, como estudios de imagen, estudio de coagulación o hemograma (en el caso que se hayan realizado); resultado del estudio histopatológico; tratamiento que recibieron y la evolución durante el seguimiento. Definimos regresión espontánea como la ausencia de infiltración superficial o profunda, aunque persistan pequeños cambios en la piel como hiperpigmentación o telangiectasias.
ResultadosEn total, recogimos 9 casos, 6 mujeres y 3 varones, con una mediana de seguimiento de 8 años (tabla 1). Según la edad de inicio, clasificamos a nuestros pacientes en pacientes con TA congénitos y adquiridos. Los TA adquiridos los dividimos en tempranos (aquellos en los que el comienzo de la clínica se produjo con una edad inferior al año de vida) y tardíos (con una edad superior al año de vida).
Angiomas en penacho diagnosticados mediante biopsia en paciente menores de 15 años en nuestro centro
| Sexo | Edad del inicio | Localización | Clínica | Malformación A-V | Manejo | Resolución espontánea | Secuelas | Complicaciones |
|---|---|---|---|---|---|---|---|---|
| 1/M | Al nacer | Miembro superior | Placa indurada eritematoviolácea | No | Vigilancia | Sí | Atrofia cutánea e hiperpigmentación | Ninguna |
| 2/V | Al nacer | Miembro superior | Macula rojo-vinosa | Sí | CE orales.Propranolol por vía oral y tópico | No | Persistencia de la lesión | Dolor, inflamación, calor local, hipertricosis, parestesias |
| 3/M | Al nacer | Dorso de pie | Tumoración eritematosa firme | Sí | No | No | Se pierde seguimiento | Se pierde seguimiento |
| 4/M | 8 años | Dorso nariz | Nódulo eritematoso indurado | No | Extirpación quirúrgica | No | No | Sangrado |
| 5/M | 10 meses | Lumbar | Tumoración blanda de aspecto vascular | No | Vigilancia | Sí | Tejido fibroadiposo residual | Ulceración |
| 6/M | Al nacer | Miembro superior | Tumoración eritematosa bilobulada elástica | No | Vigilancia | Sí | No | No |
| 7/M | Al nacer | Ingle | Tumoración violácea, indurada e infiltrada | No | Propranolol | Sí | No | No |
| 8/VV | Al nacer | Muslo | Lesión rojo-vinosa | No | Propranolol | Sí, previo a inicio propranolol | Atrofia TCS y venas prominentes | No |
| 9/V | Al nacer | Flanco | Lesión violácea semiblanda, no dolorosa, con hematoma en superficie | No | Vigilancia | Sí | No | No |
En nuestra serie encontramos 7 pacientes con TA congénitos, un paciente con TA temprano, que desarrolló la lesión a los 10 meses de vida, y un paciente con TA adquirido, con un inicio a los 9 años de edad. La localización más frecuente fue en los miembros superiores, encontrando dicha localización en 5 pacientes, seguido de tronco y muslos. La presentación clínica fue variable, predominando placas o nódulos eritematovioláceos, infiltrados, firmes, de bordes imprecisos.
Se realizó ecografía de partes blandas en 5 pacientes, observándose asociación a malformación arterio-venosa en solo un paciente, al cual se estudió mediante RM vascular, en la que se confirmó la presencia de hipervascularización y de micronódulos en tejido celular subcutáneo en relación con microfístulas arteriovenosas.
El diagnóstico definitivo en todos los casos fue mediante biopsia. En el estudio histológico con H-E se observaron múltiples lóbulos de capilares de pequeño tamaño, distribuidos en la dermis reticular en todos los casos, con afectación del panículo adiposo en 6 de ellos. Tanto en la periferia como dentro de los lóbulos angiomatosos, se observó también la presencia de vasos dilatados de paredes finas, dando lugar a espacios vasculares con forma de semiluna (fig. 1). No se observaron componentes inflamatorios en el estroma ni tampoco atipias. Se realizaron tinciones para estudio inmunohistoquímico en las biopsias de 2 de los pacientes; la tinción para GLUT-1 fue negativa, a diferencia de lo que sucede en los hemangiomas de la infancia, y la tinción para podoplanina (D2-40) fue positiva en los linfáticos del estroma y parcialmente en los capilares de dentro de los lóbulos, lo que demuestra la diferenciación parcial linfática del TA.
 Biopsia de piel (HE, 40×): proliferación de lóbulos redondeados en dermis media y profunda. B) Lóbulos de capilares que llegan hasta hipodermis (HE, 100×). C) Apariencia típica de los agregados vasculares mostrando paquetes compactos de capilares y vasos de paredes finas en semiluna tanto dentro como fuera de los lóbulos (HE, 200×). D) La tinción para podoplanina (D2-40, 200×) demuestra diferenciación parcial linfática dentro de los lóbulos de capilares.")
A) Biopsia de piel (HE, 40×): proliferación de lóbulos redondeados en dermis media y profunda. B) Lóbulos de capilares que llegan hasta hipodermis (HE, 100×). C) Apariencia típica de los agregados vasculares mostrando paquetes compactos de capilares y vasos de paredes finas en semiluna tanto dentro como fuera de los lóbulos (HE, 200×). D) La tinción para podoplanina (D2-40, 200×) demuestra diferenciación parcial linfática dentro de los lóbulos de capilares.
En cuanto a la evolución de los TA, sabemos que aquellos que tienen un origen congénito o que aparecen en edad temprana presentan mayor tendencia a la regresión que los que se originan a una edad más tardía7. En nuestra serie, observamos regresión de las lesiones en 6 pacientes; en 5 de ellos, la regresión fue espontánea y otro paciente presentó regresión con tratamiento. En un paciente la lesión se estabilizó tras iniciar tratamiento y en otro paciente se realizó extirpación quirúrgica del angioma, sin recidiva posterior.
De los 5 casos en los que se decidió abstención terapéutica y que presentaron regresión espontánea, 4 de ellos eran de origen congénito y un paciente de inicio temprano. Todos estos pacientes siguieron controles clínicos periódicos, observándose una media de inicio de la regresión espontánea a los 3,5 meses del diagnóstico, con disminución progresiva del componente profundo y de la induración a lo largo del tiempo, persistiendo en algunos de ellos discreta atrofia del tejido celular subcutáneo, telangiectasias en superficie o coloración violácea (fig. 2).
 Angioma en penacho congénito en la región lumbar de un niño. B) Tras 6 meses de seguimiento, sin tratamiento, presenta involución de la lesión con disminución del componente profundo y de la intensidad de coloración en superficie.")
En un paciente con TA congénito en la cara interna del muslo izquierdo, en el que ya se observaba cierta tendencia a la regresión, se decidió iniciar tratamiento con propranolol por vía oral a dosis de 2mg/kg/día en uso compasivo. Tras 5 meses de tratamiento, se suspendió al observar desaparición del componente profundo, apreciándose así mismo una marcada atrofia del tejido celular subcutáneo y venas prominentes, sin asociar compromiso funcional del miembro (fig. 3). No podemos atribuir la mejoría clínica del paciente ni la atrofia secundaria al tratamiento con propranolol por vía oral, ya que el paciente había iniciado involución espontánea previa al comienzo del tratamiento; además, existen casos publicados en la literatura de pacientes con secuelas similares tras ser dejados a su evolución espontánea7.
 Angioma en penacho en cara interna de muslo derecho de 10×15cm, presente desde el nacimiento, previo a tratamiento con propranolol 2mg/kg/día durante 3 meses. B) Control a los 4 años, se observa involución dejando atrofia de tejido celular subcutáneo.")
El paciente en el que se estabilizó la lesión tras tratamiento presentaba una lesión violácea indurada en cara interna de brazo izquierdo desde el nacimiento, que había presentado durante el seguimiento episodios de inflamación y dolor local. Con técnicas de imagen se demostró la presencia de malformación arterio-venosa asociada, descartándose fenómeno de Kasabach-Merritt mediante analítica. Este paciente recibió tratamiento con prednisolona a dosis de 3mg/kg/día durante varios meses, mejorando la sintomatología clínica y la inflamación. A los 9 años de edad, se decide tratamiento con propranolol por vía oral 2mg/kg/día, que el paciente suspende tras 6 meses de tratamiento por presentar intensa astenia, sin observarse mejoría clínica. La lesión continúa estabilizada en la actualidad (fig. 4).
 Angioma en penacho asociado a malformación A-V. B) Tras 5 meses de tratamiento con propranolol 2mg/kg/día, la lesión permanece estable, sin mejoría. C) Tras 3 sesiones de PDL, parámetros: spot 7mm, 0,5 ms y 9, 10 y 10,5J/cm2, con mejoría clínica y sin cicatrices.")
Ninguno de nuestros pacientes presentó el fenómeno de Kasabach-Merritt, trombocitopenia, anemia hemolítica y coagulopatía de consumo que se ha descrito en el 10% de los pacientes con TA, que puede poner en peligro la vida del paciente.
DiscusiónEl TA es una tumoración vascular poco frecuente. Su presentación clínica es muy variable; debemos sospechar que estamos ante esta entidad en un niño con lesión vascular indurada, eritematosa o purpúrica, con bordes mal definidos, aunque en ciertos casos podemos observar lesiones bien delimitadas. Suelen tener un componente nodular profundo (pueden extenderse hacia el tejido celular subcutáneo, fascia y músculo). A veces, la lesión es ligeramente dolorosa. En algunos pacientes existe hiperhidrosis e hipertricosis, o coloración a tipo mancha de Oporto en la superficie.
La patogenia del TA no está clara, se ha relacionado con un aumento de secreción a nivel local de factores de crecimiento que son importantes en la angiogénesis, como interleucina 8, que estimularían la angiogénesis, favoreciendo el desarrollo de los lóbulos de capilares8.
Para el diagnóstico es necesario combinar la clínica, la histología y los estudios de imagen, aunque el diagnóstico definitivo es histopatológico.
El diagnóstico diferencial debemos hacerlo con los hemangiomas infantiles, los hemangiomas congénitos, el hemangiopericitoma infantil, el fibroxantoma infantil, malformaciones venosas, sarcoma de Kaposi o el hemangioendotelioma kaposiforme (HEK).
El HEK tiene una apariencia purpúrica característica; es un tumor agresivo localmente, que puede afectar a tejidos profundos, incluso retroperitoneo, mediastino u órganos internos. Está totalmente aceptado que el TA y el HEK representan 2 polos finales de un mismo espectro de hallazgos histopatológicos. Típicamente, el TA es más superficial y el HEK se localiza en tejidos profundos, pudiendo, sin embargo, encontrar en un mismo tumor imágenes histológicas de los dos espectros. Además, ambos tumores se asocian a coagulopatía y/o fenómeno de Kasabach-Merritt, que puede observarse hasta en un 70% de los casos en el HEK y en el 10% de los casos de TA. Todo esto apoya la idea de que ambas entidades forman parte de un mismo espectro neoplásico9,10.
Los estudios de imagen, como la ecografía o la RM, pueden ser de ayuda para diferenciar el TA de otros tumores, aunque su principal utilidad radica en la delimitación de la extensión. También pueden servir para valorar la respuesta al tratamiento.
En caso de que estemos ante una lesión de gran tamaño, que ha aumentado rápidamente, dolorosa o con otros síntomas de alarma, es necesaria la realización de pruebas de laboratorio, como hemograma y estudio de coagulación, para descartar fenómeno de Kasabach-Merritt11.
Histopatológicamente, el TA muestra múltiples lóbulos celulares salpicados por todo el espesor de la dermis, que con frecuencia se extienden a hipodermis. Cada lóbulo está constituido por agregados de células endoteliales que se disponen concéntricamente alrededor de los vasos de los plexos vasculares de la dermis. Estos lóbulos están separados por dermis normal, sin observarse infiltrado inflamatorio. Algunos de estos lóbulos protruyen sobre las paredes vasculares circundantes, dando una morfología en semiluna característica. También podemos observar pequeños espacios vasculares de apariencia capilar en el espesor de los lóbulos de células endoteliales.
Los estudios de inmunohistoquímica demuestran que las células de los lóbulos neoplásicos presentan positividad para marcadores de células endoteliales como CD31, CD34, WT1 y actina de músculo liso. Los vasos dilatados en semiluna de la periferia se tiñen de forma variable con podoplanina (D2-40), demostrando su naturaleza linfática. Clásicamente, se pensaba que los lóbulos de capilares eran D2-40 negativos, pero en un estudio más reciente se ha observado positividad para este marcador y para PROX 1, demostrándose de esta manera la naturaleza parcial linfática del TA. Todos estos hallazgos apoyan la idea del solapamiento entre el hemangioma en penacho y el hemangioendotelioma kaposiforme, que sabemos que clásicamente es positivo para D2-4012,13.
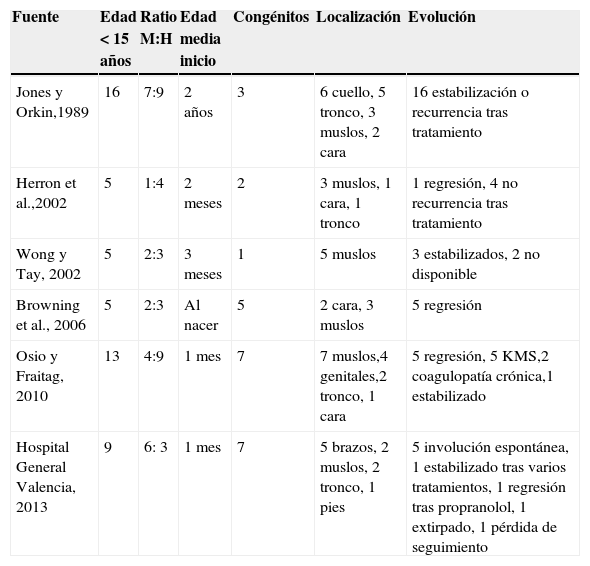
Aunque el TA puede aparecer en adultos, la mayoría de los casos aparecen en la infancia. En una revisión de 41 casos, realizada en el año 2000, más del 50% de los casos se manifestaban antes del año de vida14. La frecuencia de TA congénitos es variable en las diferentes publicaciones; en un estudio de la década de los 90, se estimó una prevalencia del 10%15; en una revisión del año 2000 se observó un 20% y en una serie de 16 pacientes del año 2010 se describió una frecuencia de TA congénitos del 54%16. En nuestra serie de 9 pacientes, la frecuencia de TA congénitos fue del 77,7%, la cual es una frecuencia bastante superior a la descrita previamente. Comparando a nuestros pacientes con el resto de series publicadas (tabla 2) 6,17,18,7, podemos observar una alta tasa de involución espontánea, probablemente atribuible al alto porcentaje de TA congénitos. Además, podemos destacar un predominio femenino y una localización más frecuente en miembros superiores (55,5%), siendo más común en otras series la afectación de muslos y cuello.
Comparación de nuestra serie con series publicadas previamente de angiomas en penacho en la infancia
| Fuente | Edad < 15 años | Ratio M:H | Edad media inicio | Congénitos | Localización | Evolución |
|---|---|---|---|---|---|---|
| Jones y Orkin,1989 | 16 | 7:9 | 2 años | 3 | 6 cuello, 5 tronco, 3 muslos, 2 cara | 16 estabilización o recurrencia tras tratamiento |
| Herron et al.,2002 | 5 | 1:4 | 2 meses | 2 | 3 muslos, 1 cara, 1 tronco | 1 regresión, 4 no recurrencia tras tratamiento |
| Wong y Tay, 2002 | 5 | 2:3 | 3 meses | 1 | 5 muslos | 3 estabilizados, 2 no disponible |
| Browning et al., 2006 | 5 | 2:3 | Al nacer | 5 | 2 cara, 3 muslos | 5 regresión |
| Osio y Fraitag, 2010 | 13 | 4:9 | 1 mes | 7 | 7 muslos,4 genitales,2 tronco, 1 cara | 5 regresión, 5 KMS,2 coagulopatía crónica,1 estabilizado |
| Hospital General Valencia, 2013 | 9 | 6: 3 | 1 mes | 7 | 5 brazos, 2 muslos, 2 tronco, 1 pies | 5 involución espontánea, 1 estabilizado tras varios tratamientos, 1 regresión tras propranolol, 1 extirpado, 1 pérdida de seguimiento |
Se han visto diferencias en cuanto a la evolución de los TA. Con frecuencia presentan una fase de crecimiento lento durante varios meses, seguido de una fase de estabilización19. En cuanto a la regresión espontánea, en una revisión de 27 casos de TA congénitos20, se observó que en el 95% la regresión espontánea ocurría en un periodo inferior a los 2 años de inicio de la clínica. En cambio, los TA adquiridos suelen presentar una fase de crecimiento lento más larga, con poca tendencia a la regresión espontánea, tan solo en el 15,9% de los casos21.
En cuanto al manejo, no existe un tratamiento estandarizado y la mayoría de los tratamientos publicados son de casos aislados o pequeñas series. El manejo debe ser individualizado, considerando la localización, el tamaño, la presencia o no de compromiso funcional y la asociación con fenómeno de Kasabach-Merritt. En cuanto al tratamiento del TA no asociado a fenómeno de Kasabach-Merritt, la abstención terapéutica y vigilancia puede ser una buena opción en lesiones no complicadas, asintomáticas y que no comprometan ningún órgano importante, ya que la probabilidad de involución espontánea es alta20. Si la lesión es de pequeño tamaño, la escisión quirúrgica es otra opción a tener en cuenta, sin embargo, debido a que los márgenes suelen ser mal definidos, la extirpación completa es difícil22,23. Se han descrito otros tratamientos con diferentes tasas de respuesta, como corticoides sistémicos a altas dosis24, interferón alfa y quimioterápicos como vincristina25,26. En los últimos, años se han obtenido buenos resultados con bajas dosis de aspirina27 asociada o no a ticoplidina, por lo que muchos autores en estos momentos consideran como primera línea de tratamiento del TA no complicado dosis de 5mg/kg/día de aspirina. Dado el éxito de la terapia con propranolol por vía oral en los hemangiomas de la infancia, se ha usado en algunos casos de AT. En una serie de 11 pacientes con HEK y AP tratados con propranolol por vía oral, solo se vio mejoría en el 36% de ellos28, con un alto índice de regresiones espontáneas, por lo que este tratamiento no puede considerarse efectivo para el TA. Por último, existen tratamientos encaminados a mejorar la apariencia estética de las lesiones, como es el caso del láser PDL o IPL, que son útiles para la mácula tipo mancha vino de Oporto superficial que tiene algunos TA.
ConclusionesPresentamos una serie de 9 pacientes diagnosticados de TA en la infancia, siendo el 77,7% de ellos congénitos y con un largo periodo de seguimiento. Nuestra serie es una de las series más largas descritas en la literatura hasta el momento. Todos nuestros pacientes fueron diagnosticados mediante biopsia, ya que dada la variabilidad clínica de esta entidad es necesario un examen histológico para el diagnóstico. Observamos regresión espontánea en el 55,5% de los pacientes, siendo la mayoría de estos congénitos. Este porcentaje de involución espontánea es superior al descrito previamente en otras series. Como conclusión, dada la alta tasa de involución espontánea en TA congénitos o tempranos, en ausencia de otras complicaciones, la vigilancia es una buena opción de manejo, siempre monitorizando estrechamente al paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
