




Tufted angioma (TA) is a rare benign vascular tumour that mostly appears during infancy or early childhood, although there are cases reported in adults. Clinical presentation and evolution of TA can vary. Histologically, it takes on a classic appearance of vascular tufts (“cannon ball” like appearance).
Patients and methodsA retrospective observational study was conducted that included all patients diagnosed with TA at our centre in the last 20 years.
ResultsA series of 9 cases of tufted angioma in childhood are presented, 77.7% of which were congenital. This represents a frequency higher than previously described. Spontaneous regression was observed in 55.5% of the cases, and was more frequent in the congenital TA group. Unlike other TA series reported in the literature, a higher proportion of patients with spontaneous regression was observed in this series, with a higher prevalence in females (6 out of 9 children) and predominantly located in the upper limbs. None of our patients had Kasabach–Merritt phenomenon.
ConclusionsThere are many ways of treating TA, but none are uniformly effective. Given the high rate of spontaneous regression in congenital or early TA, we suggest that, in the absence of other complications, monitoring would be a good option for management.
El angioma en penacho o tufted angioma (TA) es una tumoración vascular benigna poco frecuente, que suele aparecer en la infancia, aunque existen casos de aparición en la edad adulta. Su presentación clínica es muy variable. Se manifiesta típicamente como una mácula, pápula o nódulo eritematovioláceo en el tronco o el cuello. Histológicamente, se caracteriza por agregados de lóbulos angiomatosos en la dermis formando pequeños penachos de capilares.
Pacientes y métodosEstudio retrospectivo observacional de los casos diagnosticados de TA en los últimos 20 años en nuestro centro.
ResultadosPresentamos un total de 9 casos de angiomas en penacho en la infancia. El 77,7% de los casos fueron congénitos, lo que representa una frecuencia superior a la descrita previamente. Nuestros pacientes presentaron regresión espontánea en el 55,5% de los casos, siendo más frecuente en el grupo de TA congénitos. A diferencia de las otras series descritas en la literatura, observamos un mayor porcentaje de pacientes con regresión espontánea, un predominio femenino (6 de los 9 niños) y una localización más frecuente en miembros los superiores. Ninguno de nuestros pacientes presentó fenómeno de Kasabach-Merritt.
ConclusionesDada la alta tasa de involución espontánea en TA congénitos o tempranos, en ausencia de otras complicaciones, la vigilancia sería una buena opción de manejo, monitorizando estrechamente al paciente.
Tufted angioma (TA) is a rare benign vascular tumour. It was first described by Nakagawa as angioblastoma in 1949.1 Later on, in 1971, Jones and Orkin1 presented a series of cases with similar histological characteristics and coined the term “tufted angioma”. This term derives from its histological appearance, consisting of small “tufts” of capillaries and angiomatous lobules in the dermis associated with dilated lymphatic channels and with characteristic “cannonball” distribution.2 Tufted angioma may be congenital or acquired, and the incidence of congenital cases is greater than initially believed, rising to 50% in recent series. Acquired cases generally appear in the early years of life, although the literature has described cases with onset in adulthood.3,4 Although there are cases of disseminated TA,5 most present with a solitary lesion, an erythematous, purplish, infiltrated and usually poorly demarcated plaque.6 Most patients are asymptomatic, although they can experience local discomfort in the form of warmth, tenderness or swelling, and the most severe potential complication is Kasabach–Merritt syndrome.
Patients and methodsWe conducted an observational and descriptive retrospective study by reviewing the cases of TA diagnosed by biopsy in the past 20 years in the Hospital General Universitario of Valencia in children less than 15 years of age. Patients were selected by reviewing the paper medical records and the electronic records in our hospital's intranet. We excluded patients that were more than 15 years of age at the onset and patients without a histological diagnosis.
We included a total of nine patients for whom we collected epidemiological data such as sex, age at onset and ethnicity; clinical data such as localisation, appearance, and clinical diagnosis; data on diagnostic testing, such as imaging studies, coagulation tests, or complete blood counts (whenever they were performed); results from the histological examination; treatment received and outcomes during followup. We defined spontaneous regression as the absence of superficial or deep infiltration, even in the presence of residual skin changes, such as hyperpigmentation or telangiectasias.
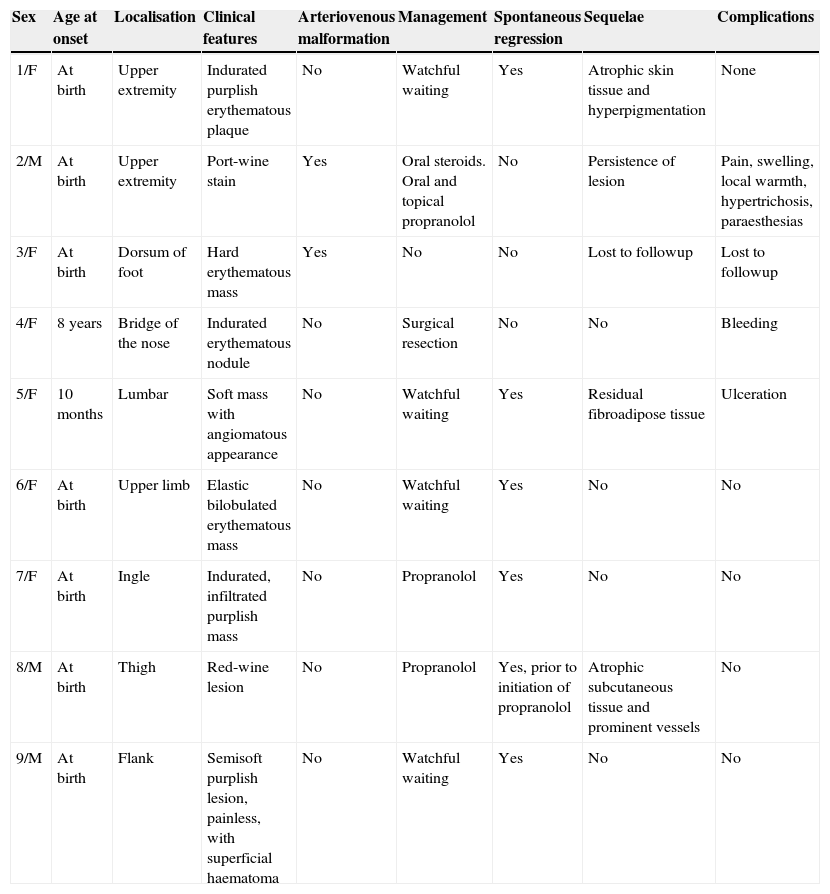
ResultsWe included a total of nine cases, six in females and three in males, with a mean followup duration of 8 years (Table 1). We classified the cases of TA as congenital or acquired based on the age at onset. We further divided acquired TAs into early-onset (onset of symptoms at age <1 year) and late-onset (at age >1 year).
Tufted angiomas diagnosed by biopsy in patients younger than 15 years at our hospital.
| Sex | Age at onset | Localisation | Clinical features | Arteriovenous malformation | Management | Spontaneous regression | Sequelae | Complications |
|---|---|---|---|---|---|---|---|---|
| 1/F | At birth | Upper extremity | Indurated purplish erythematous plaque | No | Watchful waiting | Yes | Atrophic skin tissue and hyperpigmentation | None |
| 2/M | At birth | Upper extremity | Port-wine stain | Yes | Oral steroids. Oral and topical propranolol | No | Persistence of lesion | Pain, swelling, local warmth, hypertrichosis, paraesthesias |
| 3/F | At birth | Dorsum of foot | Hard erythematous mass | Yes | No | No | Lost to followup | Lost to followup |
| 4/F | 8 years | Bridge of the nose | Indurated erythematous nodule | No | Surgical resection | No | No | Bleeding |
| 5/F | 10 months | Lumbar | Soft mass with angiomatous appearance | No | Watchful waiting | Yes | Residual fibroadipose tissue | Ulceration |
| 6/F | At birth | Upper limb | Elastic bilobulated erythematous mass | No | Watchful waiting | Yes | No | No |
| 7/F | At birth | Ingle | Indurated, infiltrated purplish mass | No | Propranolol | Yes | No | No |
| 8/M | At birth | Thigh | Red-wine lesion | No | Propranolol | Yes, prior to initiation of propranolol | Atrophic subcutaneous tissue and prominent vessels | No |
| 9/M | At birth | Flank | Semisoft purplish lesion, painless, with superficial haematoma | No | Watchful waiting | Yes | No | No |
We found seven patients with congenital TA, one patient with early-onset TA that developed the lesion at 10 months of age, and one patient with late onset at 9 years of age. The lesions most frequently localised to the upper extremities (five patients), followed by the torso and thighs. The clinical manifestations were heterogeneous, with a predominance of hard, poorly demarcated and infiltrated purplish erythematous plaques or nodules.
A soft-tissue ultrasound was performed in five patients, revealing an arteriovenous malformation in only one patient. A vascular MRI scan was performed in this patient, confirming the presence of hypervascularization and micronodules in the subcutaneous tissue associated with arteriovenous microfistulas.
In every patient, the definitive diagnosis was made based on biopsy results. The histopathological examination of haematoxylin and eosin (HE) stained sections showed multiple lobules of small capillaries that were distributed through the reticular dermis in every patient, with involvement of the subcutaneous fat layer in six. The presence of dilated thin-wall vessels that gave rise to crescent-shaped vascular channels was observed in the periphery as well as inside the vascular lobules (Fig. 1). There was no evidence of atypia or inflammatory infiltrate in the stroma. Immunohistochemical staining was performed in the biopsies of two patients; the GLUT-1 stain was negative, unlike what happens in childhood haemangiomas, while the podoplanin stain (D2-40) was positive in the lymphatic channels of the stroma and in parts of the capillaries contained in the lobules, which demonstrated the partial lymphatic differentiation found in TA.
 Skin biopsy (HE, 40×): proliferation of round lobules in middle and deep dermis. (B) Capillary lobules reaching the hypodermis (HE, 100×). (C) Typical appearance of angiomatous aggregates showing compact bundles of capillaries and thin-walled crescent-shaped vessels both in and out of the lobules (HE, 200×). (D) Podoplanin staining (D2-40, 200×) shows partial lymphatic differentiation within the capillary lobules.")
(A) Skin biopsy (HE, 40×): proliferation of round lobules in middle and deep dermis. (B) Capillary lobules reaching the hypodermis (HE, 100×). (C) Typical appearance of angiomatous aggregates showing compact bundles of capillaries and thin-walled crescent-shaped vessels both in and out of the lobules (HE, 200×). (D) Podoplanin staining (D2-40, 200×) shows partial lymphatic differentiation within the capillary lobules.
As for the outcome of TA, it is known that congenital and early-onset cases are more likely to regress than cases that appear later in life.7 In our series, we observed regression of the lesions in six patients, which was spontaneous in five and resulted from treatment in one other. In one patient, the lesion stabilised after initiating treatment, and the angioma was surgically resected in another patient, in whom it did not recur.
Of the five cases in which it was decided not to treat the patient and that showed spontaneous regression, four were congenital and one had early onset. All these patients were followed up in regular clinical checkups, and the observed mean time to the start of spontaneous regression was 3.5 months since diagnosis, with a progressive reduction of the deep component and of induration over time; in some of these patients, mild subcutaneous tissue atrophy, superficial telangiectasias or purplish colouration remained (Fig. 2).
 Congenital tufted angioma in the lumbar region of a child. (B) After 6 months of followup with no treatment, the lesion shows involution with a reduction in its deep component and in the intensity of the surface colouration.")
In one patient that had congenital TA in the inner left thigh and was starting to show some signs of regression, the decision was made to initiate compassionate treatment with oral propranolol at doses of 2mg/kg/day. After 5 months, treatment was suspended after finding that the deep component had regressed. There was also evidence of marked atrophy in the subcutaneous tissue and prominent blood vessels, with no functional compromise in the limb (Fig. 3). We cannot attribute the clinical improvement of the patient nor the secondary atrophy to the oral treatment with propranolol, as spontaneous involution had already started in the patient before treatment was initiated; furthermore, there are published cases in the literature of patients with similar sequelae following spontaneous regression under watchful waiting.7
 Tufted angioma in right inner thigh measuring 10cm×15cm, present since birth, previous to treatment with 2mg/kg/day of propanolol for 3 months. (B) Four-year followup: the lesion had regressed, leaving behind subcutaneous atrophic tissue.")
The patient in whom the lesion stabilised with treatment had a hard purplish lesion in the inner arm since birth and had experienced episodes of local pain and swelling during the followup. Imaging tests showed the presence of an arteriovenous malformation, and laboratory tests ruled out Kasabach–Merritt syndrome. The patient was treated with prednisone at a dose of 3mg/kg/day for several months, with improvement of the clinical manifestations and the swelling. At 9 years of age, treatment with oral propranolol at 2mg/kg/day was initiated, which the patient stopped taking after 6 months due to severe asthenia in the absence of clinical improvement. The lesion has remained stable to the present day (Fig. 4).
 Tufted angioma associated with arteriovenous malformation. (B) After a 5-month course of 2mg/kg/day of propanolol, the lesion remained unchanged, with no improvement. (C) Parameters after three PDL sessions: spot 7mm, 0.5ms and 9, 10 and 10.5J/cm2, with clinical improvement and no scars.")
(A) Tufted angioma associated with arteriovenous malformation. (B) After a 5-month course of 2mg/kg/day of propanolol, the lesion remained unchanged, with no improvement. (C) Parameters after three PDL sessions: spot 7mm, 0.5ms and 9, 10 and 10.5J/cm2, with clinical improvement and no scars.
None of our patients had Kasabach–Merritt syndrome, thrombocytopaenia, haemolytic anaemia or disseminated intravascular coagulation, which have been described in 10% of patients with TA and can be life-threatening.
DiscussionTufted angioma is a rare vascular tumour. It has a heterogeneous clinical presentation. This entity should be suspected in patients presenting with an indurated, erythematous or purplish lesion with poorly defined borders, although in some cases the lesion is well demarcated. They usually have a deep nodular component (they may extend into the subcutaneous tissues, fascia and muscle). The lesion can sometimes cause mild pain. Some patients have hyperhidrosis and hypertrichosis, or port-wine stains on the surface of the skin.
The pathogenesis of TA remains unclear. It has been associated to increased local secretion of growth factors with a significant role in angiogenesis, such as interleukin 8, which would stimulate angiogenesis and promote the development of vascular lobules.8
The diagnosis of TA is made by clinical assessment, histopathological examination and imaging tests, although the definitive diagnosis is given by the histopathological examination.
The differential diagnosis must include childhood haemangiomas, congenital haemangiomas, childhood haemangiopericytoma, childhood fibroxanthoma, venous malformations, Kaposi's sarcoma and kaposiform haemangioendothelioma (KHE).
Kaposiform haemangioendothelioma has a characteristic purpura-like appearance; it is a locally aggressive tumour that may affect deep tissues, even the retroperitoneum, mediastinum or internal organs. There is consensus that TA and KHE represent the two extremes of a single spectrum of histopathological findings. While TA tends to be more superficial and KHE to localise to deep tissues, the histological patterns of both entities may be found in a single mass. Furthermore, both have an association with coagulopathy and Kasabach–Merritt syndrome, which may be found in up to 70% of KHE cases and 10% of TA cases. All of the above support the notion that both entities are part of a single neoplastic spectrum.9,10
Imaging studies such as ultrasonography or magnetic resonance imaging can help distinguish TA from other tumours, although their main contribution is determining the extension of the lesion. They can also be used to assess response to treatment.
If a patient has a large lesion that has increased in size rapidly, is painful, or is associated with other warning signs, laboratory tests such as a complete blood count and coagulations tests should be done to rule out Kasabach–Merritt syndrome.11
The histological appearance of TA consists of multiple cellular lobules scattered through the entire thickness of the dermis and that frequently extend to the hypodermis. Each lobule is composed of several aggregates of endothelial cells arranged concentrically around the vessels of the vascular plexus of the skin. These lobules are separated by normal dermis, with no evidence of inflammatory infiltrate. Some of these lobules push against the walls of surrounding vessels, producing the characteristic crescent-shaped morphology. Small vascular spaces with a capillary appearance can also be observed within the endothelial cell lobules.
Immunohistochemical tests show that the cells contained in the neoplastic lobules are positive for endothelial cell markers such as CD31, CD34, WT1 and alpha-smooth muscle actin. The crescent-shaped dilated vascular channels in the periphery are stained to different degrees by podoplanin (D2-40), which evinces their lymphatic nature. Historically it was believed that the vascular lobules were D2-40-negative, but a more recent study has shown that they test positive for this marker and for PROX 1, which is evidence of the partial lymphatic nature of TA. All of these findings support the notion of an overlap between TA and KHE, the latter of which is known to test positive for D2-40.12,13
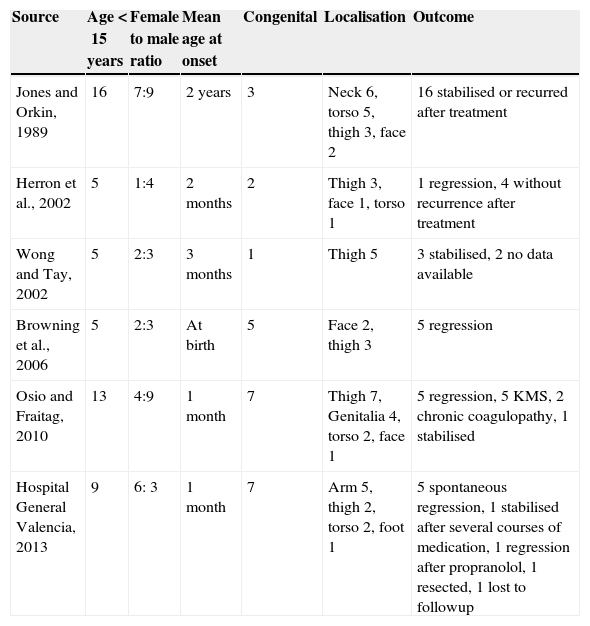
Although TA can develop in adults, most cases manifest in childhood. A review of 41 cases conducted in 2000 showed that onset occurred before age 1 year in 50% of patients.14 The reported prevalence of congenital TA varies in different publications; a study conducted in the 1990s estimated a 10% prevalence;15 a case review from 2000 found a prevalence of 20%, and the prevalence of congenital TA in a 2010 series of 16 patients was 54%.16 The frequency of congenital TA in our nine-patient series was 77.7%, which is significantly higher than the prevalences previously reported in the literature. When we compared our patients to the rest of the published series (Table 2)6,7,17,18 we observed a high rate of spontaneous regression, which could probably be attributed to the high percentage of congenital cases. Other factors worth noting in our series were the higher prevalence in the female sex and the predominant localisation in the upper extremities (55.5%), compared to other series in which thigh and neck involvement are more common.
Comparison of our series with previously published case series of childhood tufted angioma.
| Source | Age<15 years | Female to male ratio | Mean age at onset | Congenital | Localisation | Outcome |
|---|---|---|---|---|---|---|
| Jones and Orkin, 1989 | 16 | 7:9 | 2 years | 3 | Neck 6, torso 5, thigh 3, face 2 | 16 stabilised or recurred after treatment |
| Herron et al., 2002 | 5 | 1:4 | 2 months | 2 | Thigh 3, face 1, torso 1 | 1 regression, 4 without recurrence after treatment |
| Wong and Tay, 2002 | 5 | 2:3 | 3 months | 1 | Thigh 5 | 3 stabilised, 2 no data available |
| Browning et al., 2006 | 5 | 2:3 | At birth | 5 | Face 2, thigh 3 | 5 regression |
| Osio and Fraitag, 2010 | 13 | 4:9 | 1 month | 7 | Thigh 7, Genitalia 4, torso 2, face 1 | 5 regression, 5 KMS, 2 chronic coagulopathy, 1 stabilised |
| Hospital General Valencia, 2013 | 9 | 6: 3 | 1 month | 7 | Arm 5, thigh 2, torso 2, foot 1 | 5 spontaneous regression, 1 stabilised after several courses of medication, 1 regression after propranolol, 1 resected, 1 lost to followup |
We found differences in the disease course. Tufted angiomas frequently show a slow growth phase that lasts a few months, followed by a stabilisation phase.19 As for spontaneous regression, a review of 27 cases of congenital TA20 found that in 95% spontaneous regression occurred less than 2 years since the onset of symptoms. Meanwhile, acquired TAs tend to have a longer slow-growth phase and are not likely to spontaneously regress, which happens in only 15.9% of cases.21
When it comes to the management of TA, it has not been standardised, and most publications that refer to its treatment consist of single-case reports or small case series. The management should be determined on a case-to-case basis, taking into account localisation, size, presence or absence of functional compromise, and comorbidity with Kasabach–Merritt syndrome. Watchful waiting without treatment may be a good strategy in patients with TA that do not have Kasabach–Merritt syndrome and have uncomplicated and asymptomatic lesions that do not involve a vital organ, as there is a high probability of spontaneous regression.20 Surgical resection should be considered if the lesion is small, however, complete resection may prove challenging given that the lesions are usually not well-demarcated.22,23 Other treatments have been reported in the literature with varying degrees of success, such as high-dose systemic corticosteroids,24 interferon alpha, and chemotherapy medications such as vincristine.25,26 In recent years, good outcomes have resulted from treatment with low-dose aspirin27 alone or in combination with ticlopidine, so many authors now propose doses of 5mg/kg/day of aspirin for the first-line treatment of uncomplicated TA. Given the success rate of treatment of childhood haemangiomas with oral propranolol, this agent has been used in some cases of TA. A series of 11 patients with KHE and TA treated with oral propranolol showed improvement in only 36% of the patients,28 and there was a high rate of spontaneous regression, so this treatment cannot be considered effective for TA. Last of all, there are treatments that aim to improve the aesthetic appearance of the lesions, such as laser treatment with PDL or IPL, which are useful for the superficial port-wine stains found in some TAs.
ConclusionsWe present a series of nine patients diagnosed with TA in childhood, in which 77.7% of the cases were congenital and subject to a long followup period. Our series is one of the longest described in the literature to date. All our patients were diagnosed by biopsy, since the clinical heterogeneity of this entity requires that the diagnosis be made by histopathological examination. We observed spontaneous regression in 55.5% of patients, most of whom had congenital TAs. This percentage of spontaneous involution was higher than percentages previously reported in other case series. In conclusion, given the high rate of spontaneous regression in congenital and early-onset cases, in the absence of other complications watchful waiting is an appropriate strategy in the management of patients with TA as long as they are monitored closely.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Victoria Martínez AM, Cubells Sánchez L, Esteve Martínez A, Estela Cubells JR, Febrer Bosch I, Alegre de Miquel V, et al. Angiomas en penacho en la infancia. Serie de 9 casos yrevisión de la literatura. An Pediatr (Barc). 2015;83:201–208.
