






West syndrome (WS) is an age-dependent epileptic encephalopathy in which the prognosis varies according to the, not always identified, underlying origin.
ObjectivesTo define the profile of cryptogenic (a least studied isolated sub-group) WS, in Spain. To study its outcome, response to different treatments, and to establish prognostic factors.
Patients and methodsThe study included a review of the medical records of 16 patients diagnosed with cryptogenic WS during the period, 2000–2015. The mean follow-up time was 6.6 years, with a minimum of 2 years.
ResultsThe large majority (11/16) were male. The mean age at onset was 6 months, and 6/16 had a family history of idiopathic epilepsy. The first line treatment with vigabatrin had an electrical–clinical response in 5/16 patients, with the remaining cases responding to adrenocorticotropic hormone (ACTH). Almost half (44%) of the patients progressed to other types of epilepsy, with no difference between those treated with vigabatrin or ACTH. A greater number of adverse effects were obtained with ACTH, with no retinal involvement being observed with vigabatrin. The aetiological cause was found in 2/16. Being female, late onset, and early control of the hypsarrhythmia, were factors of a good prognosis.
ConclusionsThe overall prognosis of cryptogenic WS was more serious than expected. Although the incidence of Lennox–Gastaut syndrome was low, the progression to focal epilepsy was the most common, with it appearing within the first 2 years of the diagnosis. The initial response to vigabatrin was lower than expected, but the long-term result was comparable to ACTH.
El síndrome de West (SW) es una encefalopatía epiléptica dependiente de la edad con pronóstico variable según la etiología subyacente, no siempre identificada.
ObjetivosDefinir el perfil del SW criptogénico en nuestro medio, subgrupo menos estudiado de forma aislada. Estudiar su evolución, respuesta a los distintos tratamientos y establecer factores pronósticos.
Pacientes y métodosRevisión de historias clínicas de 16 pacientes diagnosticados de SW criptogénico durante el período 2000-2015. El tiempo de seguimiento medio fue 6,6 años y mínimo de 2 años.
Resultados11 de 16 fueron varones, la edad media de inicio fue de 6 meses y 6/16 presentaban antecedente familiar de epilepsia idiopática. El tratamiento de primera línea con vigabatrina tuvo respuesta electroclínica en 5/16 pacientes, respondiendo los casos restantes a hormona adrenocorticotropa (ACTH). El 44% de los pacientes evolucionaron a otras epilepsias, sin diferencia entre los tratados con vigabatrina o ACTH. Se obtuvo un mayor número de efectos adversos con la ACTH, no se evidenció afectación retiniana con la vigabatrina. Durante el seguimiento se llegó a la causa etiológica en 2/16. El sexo femenino, el comienzo tardío y el control precoz de la hipsarritmia resultaron factores de buen pronóstico.
ConclusionesEl pronóstico global del SW criptogénico resultó más grave de los esperado. Aunque la incidencia de síndrome de Lennox-Gastaut fue baja, la epilepsia focal resultó la evolución más frecuente apareciendo en los 2 primeros años del diagnóstico. La respuesta inicial a vigabatrina fue menor a la esperada, pero el resultado a largo plazo resultó superponible a la ACTH.
West syndrome (WS) is an age-dependent epileptic encephalopathy characterised by the triad of (1) infantile spasms, usually clustered; (2) evidence of hypsarrhythmia in the electroencephalogram (EEG), with tracings showing a chaotic, multifocal and bilateral pattern of high-amplitude waves; and (3) regression of psychomotor development, although the latter may be absent.1,2
Its estimated incidence ranges from 2 to 4 in 10000 individuals depending on the series, and therefore it is the most frequent form of epilepsy in the first year of life outside of neonatal and febrile seizures.1,3
Due to the numerous underlying causes and the variability in neurologic development in these patients, it is often categorised into different diagnostic groups. The most common classification continues to be the one proposed by the International League Against Epilepsy (ILAE), which categorises infantile spasms as symptomatic or cryptogenic.4
The symptomatic category corresponds to patients with an obvious underlying cause and/or a history of developmental delay or epileptic seizures prior to the onset of spasms, and accounts for approximately 80% of all patients with WS. Patients in whom an underlying condition is suspected but has not been identified after the aetiological investigation are categorised as having cryptogenic WS and account for the remaining 20% of cases. In the cryptogenic group, patients have normal psychomotor development at onset that goes on to deteriorate progressively in most cases. However, in a minority of cases, approximately 5% of patients with infantile spasms, the outcomes are favourable, with full resolution of the spasms and normal psychomotor development, which has led some authors to consider it a separate aetiological group labelled idiopathic,5 although this category has yet to be adopted by the ILAE. This aetiological category is based on disease outcomes, so it can only be applied through the followup of patients.
Since developmental delays present at diagnosis could be explained by both the underlying disease and the epileptic encephalopathy itself (spasms that were not detected previously or presence of hypsarrhythmia prior to onset), some authors propose abandoning the distinction between symptomatic and cryptogenic WS in favour of a classification based on the development preceding onset. In the United Kingdom Infantile Spasms Study (UKISS), 61% of patients had proven aetiology, 33% had no identified aetiology, and 6% had not been fully investigated.4 A recent study of the ILAE proposes replacing these terms with 3 new aetiologic groups: genetic, structural-metabolic and unknown.6
When it comes to the aetiological diagnosis of WS, neuroimaging is the method that offers the highest yield. The underlying cause is identified in 70% of the patients after an adequate history-taking and physical examination and magnetic resonance imaging of the head.7 Several studies have evaluated the yield of other diagnostic tests, such as genetic and metabolic investigations.6,8,9 They conclude that in children without an aetiological diagnosis after the initial investigation, a cost-efficient strategy would be performance of microarray-based comparative genomic hybridisation (aCGH) followed by an epilepsy gene panel and a general metabolic study (lactate, pyruvate, amino acids and organic acids in urine) in those patients in which the results of aCGH are inconclusive. It is estimated that this approach achieves aetiological diagnosis in an additional 10%–15% of cases. The study conducted by the National Infantile Spasms Consortium of the United States found that performance of aCGH resulted in an aetiological diagnosis in 11% of the patients that did not previously have one, the epilepsy gene panel in 31% of those patients, and the metabolic study in 4.5%.
Advances in diagnostic techniques and the interest in the underlying cause on account of its prognostic value have led to a decrease in the frequency of cryptogenic cases with an associated increase in symptomatic cases.
Infantile spasms are refractory to most anticonvulsant drugs, and only 2 drugs have clearly proven efficacious in WS, vigabatrin (VGB) and adrenocorticotropic hormone (ACTH). At present, as the most recent Cochrane review emphasises, the existing evidence is insufficient to determine the superiority of either drug for use as first-line treatment. Some studies have shown that ACTH therapy is more effective in controlling the spasms in the short-term, although its superiority in the long term has yet to be demonstrated.10–12
There are numerous studies and reviews that focus on WS overall, with particular emphasis on the symptomatic form on account of its higher prevalence. However, few studies have specifically analysed the cryptogenic form, and consequently it is less well understood and its prognosis unclear.
The aim of this study was to define the profile of cryptogenic WS in our region, exploring the clinical characteristics of the cases, the diagnostic tests performed and the subsequent outcomes. Based on these data, we identified potential prognostic factors for the purpose of improving outcomes and counselling families.
Patients and methodsWe conducted a retrospective observational study by reviewing the electronic health records of 16 patients. The statistical analysis was solely descriptive, and we did not apply any inferential methods. The inclusion criteria were: cryptogenic aetiology (understood as normal psychomotor development prior to diagnosis), no focal signs in the neurologic evaluation at diagnosis, no history of seizures prior to onset of spasms (excluding neonatal and febrile seizures), normal neuroimaging findings and lack of an attributable aetiology; diagnosis made in the 2000–2015 period; and treatment and followup in our hospital. The mean duration of followup was 6.6 years, with a minimum of 2 years and a maximum 15 years. There were no losses to followup.
We created a data collection form that included the following variables: sex, age at onset, history (obstetric, personal and family), psychomotor development at onset, epileptic seizures prior to onset of spasms, type of spasms, EEG abnormalities, delay in treatment initiation, initial treatment, response to initial treatment, persistence of spasms/hypsarrhythmia, received treatments, adverse events, relapse, psychomotor development during followup, development of other forms of epilepsy, seizure-free time and development of neurologic disease.
In adherence with the treatment protocol of our hospital, all patients received escalating doses of VGB (up to 200mg/kg/day) as first-line treatment. If VGB monotherapy proved ineffective after 2 weeks (persistence of spasms and/or hypsarrhythmia), we added a daily dose of ACTH for 14 days that was tapered off over an additional 5 weeks.
We defined unfavourable outcome as the subsequent development of other forms of epilepsy and/or the development of moderate to severe cognitive impairment based on a neuropsychiatric evaluation.
ResultsWe studied a total of 16 patients, and there were no losses during the followup period.
When it came to sex, we found a predominance of the male sex, as 11 out of the 16 patients (69%) were male. The age at onset ranged between 1.5 and 10 months, with a predominance of patients in the 5–7 months group (50%). The mean age at presentation of spasms was 6 months.
As for the medical history, 6 of the 16 patients had a family history of epilepsy of some form, all of them benign idiopathic forms in first- or second-degree relatives, and 2 had a personal history of seizures prior to onset of spasms (neonatal seizures in one and febrile seizures in the other).
The most frequent type of spasms were extensor spasms (50%), followed by flexor spasms (43%) and mixed spasms (7%). Only 4 of the patients had mild developmental delay at onset of spasms.
The time elapsed between onset of spasms and initiation of treatment was less than 1 week in 50% of patients. Per the treatment protocol of the department, all patients received VGB as initial therapy (Fig. 1).
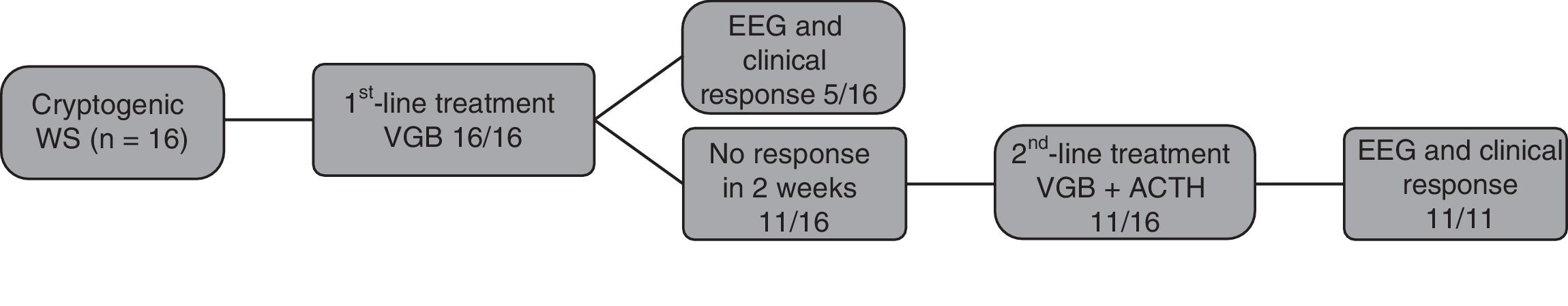
The response to VGB was favourable in 5 of the 16 patients, with resolution of the spasms in the first week of treatment and normalisation of EEG features in 2 weeks. Patients that had not improved after 15 days of VGB received ACTH as second-line treatment. All of the 11 patients treated with ACTH had a favourable response, achieving control of hypsarrhythmia.
There were adverse events in 32% of patients, most of them associated with use of ACTH. Of the 16 patients treated with VGB, only 1 suffered an adverse reaction, an acute encephalopathy that improved a few hours after discontinuing the drug. Of the 11 patients treated with ACTH, 4 developed high blood pressure requiring antihypertensive therapy; 2 of them also had echocardiographic abnormalities indicative of septal hypertrophy, although this state was transient and resolved after completion of ACTH therapy.
The outcome was unfavourable in 7 of the 16 patients (43.7%), all of who developed other forms of epilepsy. Five of them also had moderate or severe cognitive impairment, which was associated with a pervasive developmental disorder in 2. The mean time they remained seizure-free until the development of another form of epilepsy was 10 months, and secondary epilepsy developed within 2 years of diagnosis in all. Other patients exhibited milder impairment in the long term: language disorder, attention-deficit hyperactivity disorder and mild cognitive impairment, and while we have included these data in the analysis of outcomes, we did not include these patients in the unfavourable outcome group because these problems did not have a significant impact on their everyday functioning. None of the patients experienced a relapse (Table 1).
Outcomes.
| Poor outcome | n=7 |
| Secondary epilepsy | 7/7 |
| Focal epilepsy | 4 |
| Generalised epilepsy | 2 |
| Lennox–Gastaut syndrome | 1 |
| Refractory epilepsy | 2 |
| Moderate/severe cognitive impairment | 5/7 |
| Associated with epilepsy | 5 |
| Pervasive developmental disorder | 2 |
| Aetiological diagnosis found | 2/7 |
| Minor impairment | n=4 |
| ADHD | 2 |
| Language disorder | 1 |
| Mild cognitive impairment | 1 |
ADHD, attention-deficit hyperactivity disorder; n, absolute frequency.
An aetiological diagnosis was eventually achieved in 2 of the 16 patients during the followup. In one, it was biotinidase deficiency, identified when the metabolic evaluation was expanded in this patient due to the presence of persistent dermatitis, and the other a focal cortical dysplasia identified 10 years after diagnosis of spasms; both patients belonged to the unfavourable outcome group. Therefore, the overall outcome of our cryptogenic patients would improve if we were to exclude these 2 patients (who eventually became part of the symptomatic group) from the results, with unfavourable outcomes in only 5 of a total of 14 patients (35.7%).
We made a descriptive analysis of several variables, searching for potential prognostic factors with the purpose of developing working hypotheses for future inferential research. We found a greater probability of developing secondary epilepsy in patients of the male sex (55%) or onset before age 6 months (71%), while early treatment (initiated in the first 2 weeks from onset of spasms) and early control of hypsarrhythmia (in the first 3 weeks) were associated with a decreased probability of developing secondary epilepsy (36% and 33% respectively) (Table 2).
Potential prognostic factors.
| Factor | Secondary epilepsy | |
|---|---|---|
| Male sex (n=11) | 55% | 6/11 |
| Female sex (n=5) | 20% | 1/5 |
| Onset before age 6 months (n=7) | 71% | 5/7 |
| Onset after age 6 months (n=9) | 22% | 2/9 |
| With FHx of epilepsy (n=6) | 33% | 2/6 |
| Without FHx of epilepsy (n=10) | 50% | 5/10 |
| Treatment with VGB in monotherapy (n=5) | 40% | 2/5 |
| Treatment with VGB and ACTH (n=11) | 45% | 5/11 |
| Delay in treatment>2 weeks (n=5) | 60% | 3/5 |
| Delay in treatment<2 weeks (n=11) | 36% | 4/11 |
| Persistence of hypsarrhythmia>3 weeks (n=4) | 75% | 3/4 |
| Persistence of hypsarrhythmia<3 weeks | 33% | 4/11 |
ACTH, adrenocorticotropic hormone; FHx, family history; n, absolute frequency; VGB, vigabatrin.
Our study was consistent with the previous literature as regards the age of onset of spasms, the predominance of the male sex and the distribution of the type of spasms.
When we analysed the cryptogenic subgroup in particular, our study did find a higher proportion of patients with a family history of epilepsy. In previous studies on WS, the overall proportion of patients with a family history of epilepsy ranged between 15% and 25%,1,3 while in our series of cryptogenic cases it was as high as 37%. This finding is congruent with the importance of genetic factors in this group of patients, something that has already been noted in previous reviews that found an association between WS of unknown aetiology with other idiopathic forms of epilepsy, such as childhood absence epilepsy.5
Several studies have found a clear relationship between early initiation of treatment and favourable outcomes, mainly in cryptogenic cases.9,13–17 In our study, 50% of patients were referred to our hospital early, and treatment was initiated in the first week since onset of spasms. In 5 patients, treatment was initiated more than 2 weeks after onset, and 3 of them (60%) had unfavourable outcomes with subsequent development of other forms of epilepsy, which also represented a worse outcome relative to the overall outcomes of WS of any type. However, of the 11 patients that started treatment within 2 weeks of onset, only 4 (36%) developed secondary epilepsy.
Since Chiron et al.18 demonstrated the efficacy of VGB for control of spasms, numerous authors have compared VGB with ACTH and observed a similar response in the long term, although with a lower rate of adverse events.11,12,19 As we already mentioned, the treatment protocol of our department, based on the European survey study conducted by Aicardi,20–22 uses VGB monotherapy as first-line treatment and reserves ACTH therapy for patients that did not respond to the initial treatment.
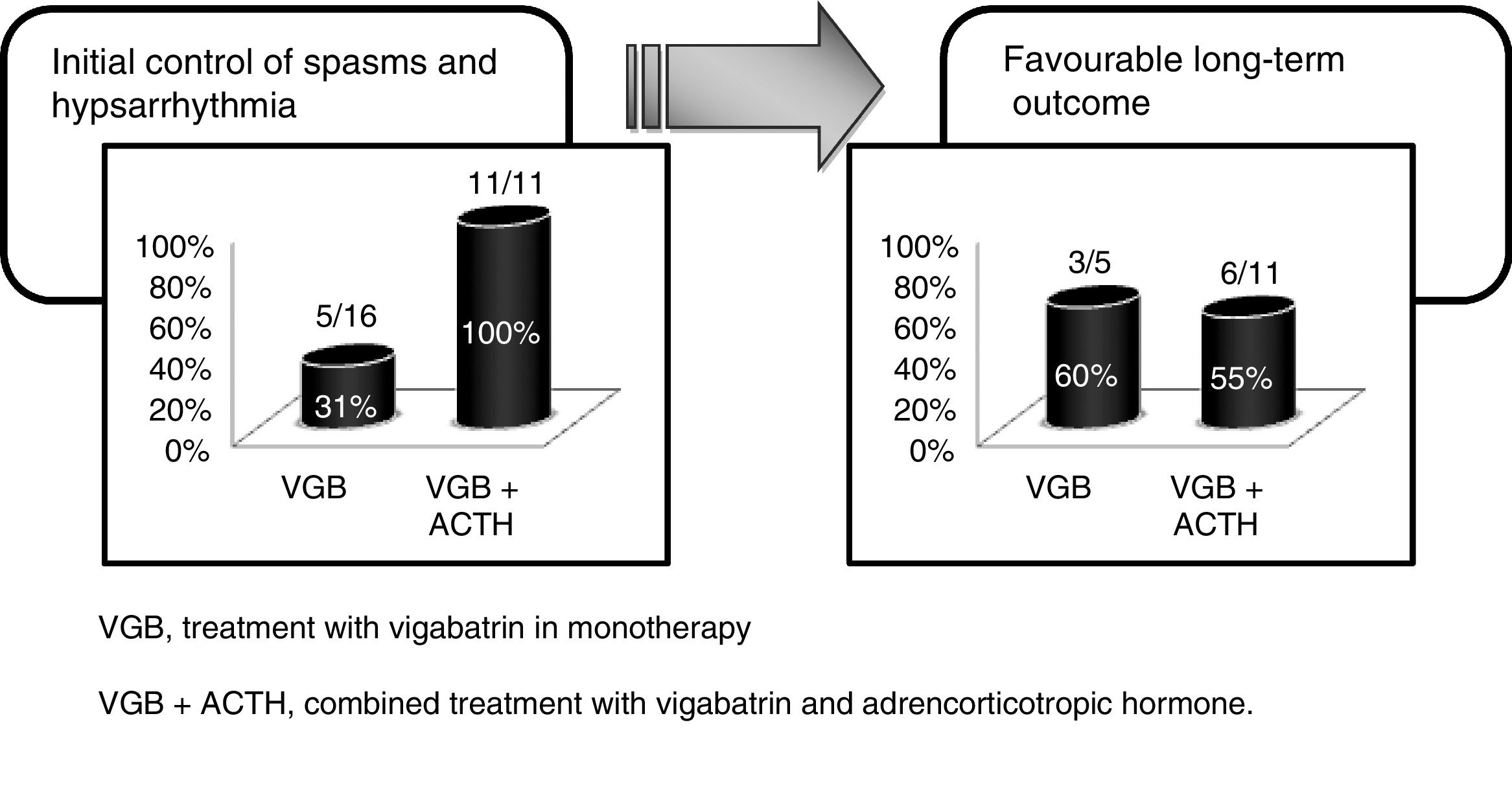
In our study, we defined a favourable response to initial therapy as resolution of spasms and normalisation of the EEG in the first 2 weeks of treatment as long as the improvement persisted for at least one month. Initial therapy with VGB achieved a favourable response in 30% of the patients with cryptogenic WS in our study, a proportion that was lower than the 50%–70% reported in other case series.1,19,23 Of the patients that responded well to initial treatment with VGB monotherapy, 60% remained free of seizures during the subsequent followup, a percentage that was similar to the percentage we found in the long-term followup of patients treated with VGB and ACTH. Therefore, while the response to initial treatment with VGB was less favourable than expected, long-term outcomes were favourable in the 56% of patients regardless of treatment, which was consistent with the findings of other studies9,10,14 (Fig. 2).
GABAergic retinopathy, which causes irreversible peripheral visual field constriction in adults, continues to be the most important adverse effect of treatment with VGB, although it is difficult to identify in the paediatric age group. Different studies have found a direct association with the dose and duration of treatment, and indicated that treatment can be safe if it lasts less than 6 months and at doses not exceeding 200mg/kg/day.15,24,25 Of the 16 patients we followed up, none experienced this complication, even though most of them received treatment for much longer, for a maximum of 3 years. On the other hand, treatment with ACTH produced moderate adverse effects that required treatment in 36% of cases.
Monitoring of retinal toxicity proved challenging due to the young age and the intellectual disability of some of the patients, although their families did not report any visual problems in their everyday lives during the followup visits. All patients underwent periodic checkups in the ophthalmology department with gross assessment of the visual field (menace response, kinetic perimetry and confrontation visual field testing) that revealed no signs of impairment. Automated perimetry was only performed in 2 patients, both without intellectual disability and once they were of prepubertal age, with results within the normal range in both, which leads us to believe that they did not suffer retinal toxicity or else the latter was reversible.
Therefore, contrary to the suggestion of other authors that advocate for first-line treatment with ACTH,26 our study did not find poorer long-term outcomes in patients treated solely with VGB, and the safety profile of this drug was adequate.
The prognosis of cryptogenic WS continues to be bleak. In our study, 44% percent of patients developed another form of epilepsy (in 71% of cases associated with moderate-to-severe cognitive impairment), findings that were pretty congruent with those of other authors.9,10,14 The incidence of Lennox–Gastaut syndrome was low (1/16), and focal epilepsy was the most frequent outcome. The patient that developed Lennox–Gastaut syndrome did not respond to monotherapy with VGB and required the addition of ACTH, in spite of which the time elapsed from initiation of treatment to resolution of hypsarrhythmia was 3 months and a half. Thus, although treatment was initiated in the first week since onset of the spasms, the persistence of hypsarrhythmia in an immature brain may have contributed to the less favourable outcome.14,27,28
Since our study only included patients with a cryptogenic aetiology, it was reasonable to expect better long-term outcomes. However, the proportion of favourable outcomes barely reached 55%, which was probably due to the presence in some cases of an unidentified underlying organic cause that worsened the prognosis independently of the control of hypsarrhythmia. Advances in diagnostic techniques and the increased use of genetic testing will lead to the identification of additional underlying conditions that could account for this epileptic encephalopathy, which a more accurate identification of cryptogenic patients that would be associated with a greater proportion of favourable long-term outcomes.6,29,30 In our case, due to lack of resources in our hospital during the period under study, genetic testing was limited to karyotype analysis and testing for microdeletion syndromes, without access to gene panels. Nevertheless, metabolic studies and neuroimaging allowed the identification of an underlying cause in 2 of the 16 patients, and excluding these patients from the analysis would increase the proportion of patients with a favourable outcome to 64%, which is closer to our original expectations.
We are aware of the limitations of our study, which is the reason we urge for the performance of additional research on this subject. Some of the methodological aspects that could be improved are the small sample size, the restriction of the analysis to descriptive methods, the limited access to diagnostic techniques that could help discriminate cryptogenic cases more accurately, and the subjective assessment of retinal toxicity.
In conclusion, although our series was limited to cases originally classified as cryptogenic, we found that a considerable percentage had unfavourable outcomes in the long term, which underscores the severity of this disease. An exhaustive aetiological investigation continues to be essential in patients with WS in order to accurately identify patients in the cryptogenic group and be able to make an accurate prognosis. In our study, the factors we identified that may be associated with a favourable prognosis were female sex, onset of spasms after age 6 months, early initiation of treatment and early control of hypsarrhythmia, with the last 2 being particularly relevant, as they are the only factors that are actually modifiable. However, we did not find an association between first-line treatment with VGB monotherapy and a greater probability of developing secondary epilepsy.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Calderón Romero M, Arce Portillo E, López Lobato M, Muñoz Cabello B, Blanco Martínez B, Madruga Garrido M, et al. Síndrome de West criptogénico: perfil clínico, respuesta al tratamiento y factores pronósticos. An Pediatr (Barc). 2018;89:176–182.
Previous presentation: Partial results of this study were presented in poster format at the XXXIX Meeting of the Sociedad Espanola de Neurología Pediátrica, May19–21, 2016; Toledo, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals