



El síndrome de West (SW) es una encefalopatía epiléptica dependiente de la edad con pronóstico variable según la etiología subyacente, no siempre identificada.
ObjetivosDefinir el perfil del SW criptogénico en nuestro medio, subgrupo menos estudiado de forma aislada. Estudiar su evolución, respuesta a los distintos tratamientos y establecer factores pronósticos.
Pacientes y métodosRevisión de historias clínicas de 16 pacientes diagnosticados de SW criptogénico durante el período 2000-2015. El tiempo de seguimiento medio fue 6,6 años y mínimo de 2 años.
Resultados11 de 16 fueron varones, la edad media de inicio fue de 6 meses y 6/16 presentaban antecedente familiar de epilepsia idiopática. El tratamiento de primera línea con vigabatrina tuvo respuesta electroclínica en 5/16 pacientes, respondiendo los casos restantes a hormona adrenocorticotropa (ACTH). El 44% de los pacientes evolucionaron a otras epilepsias, sin diferencia entre los tratados con vigabatrina o ACTH. Se obtuvo un mayor número de efectos adversos con la ACTH, no se evidenció afectación retiniana con la vigabatrina. Durante el seguimiento se llegó a la causa etiológica en 2/16. El sexo femenino, el comienzo tardío y el control precoz de la hipsarritmia resultaron factores de buen pronóstico.
ConclusionesEl pronóstico global del SW criptogénico resultó más grave de los esperado. Aunque la incidencia de síndrome de Lennox-Gastaut fue baja, la epilepsia focal resultó la evolución más frecuente apareciendo en los 2 primeros años del diagnóstico. La respuesta inicial a vigabatrina fue menor a la esperada, pero el resultado a largo plazo resultó superponible a la ACTH.
West syndrome (WS) is an age-dependent epileptic encephalopathy in which the prognosis varies according to the, not always identified, underlying origin.
ObjectivesTo define the profile of cryptogenic (a least studied isolated sub-group) WS, in Spain. To study its outcome, response to different treatments, and to establish prognostic factors.
Patients and methodsThe study included a review of the medical records of 16 patients diagnosed with cryptogenic WS during the period, 2000-2015. The mean follow-up time was 6.6 years, with a minimum of 2 years.
ResultsThe large majority (11/16) were male. The mean age at onset was 6 months, and 6/16 had a family history of idiopathic epilepsy. The first line treatment with vigabatrin had an electrical-clinical response in 5/16 patients, with the remaining cases responding to adrenocorticotropic hormone (ACTH). Almost half (44%) of the patients progressed to other types of epilepsy, with no difference between those treated with vigabatrin or ACTH. A greater number of adverse effects were obtained with ACTH, with no retinal involvement being observed with vigabatrin. The aetiological cause was found in 2/16. Being female, late onset, and early control of the hypsarrhythmia, were factors of a good prognosis.
ConclusionsThe overall prognosis of cryptogenic WS was more serious than expected. Although the incidence of Lennox-Gastaut syndrome was low, the progression to focal epilepsy was the most common, with it appearing within the first 2 years of the diagnosis. The initial response to vigabatrin was lower than expected, but the long-term result was comparable to ACTH.
El síndrome de West (SW) es una encefalopatía epiléptica dependiente de la edad caracterizada por la triada electroclínica de (1)espasmos infantiles, que típicamente ocurren en clúster; (2) la presencia de hipsarritmia en el electroencefalograma (EEG), con trazado caótico de alto voltaje, localización difusa y bilateral; (3) y detención en el desarrollo psicomotor, pudiendo estar este último ausente1,2.
La incidencia estimada varía de 2 a 4/10.000 según las series disponibles, representando por tanto la forma más frecuente de epilepsia en el primer año de vida, excluidas las convulsiones neonatales y crisis febriles1,3.
Debido a las numerosas causas subyacentes y al desarrollo neurológico variable en estos pacientes, es común su clasificación en grupos diagnósticos. La nomenclatura más usada sigue la propuesta de la International League Against Epilepsy (ILAE), agrupando los espasmos infantiles en sintomáticos y criptogénicos4.
El término sintomático hace referencia a aquellos pacientes con una clara causa subyacente y/o presencia de retrasos psicomotor o crisis epilépticas previas a la aparición de los espasmos, lo que representa aproximadamente el 80% de los pacientes con SW. Aquellos pacientes en los que se presupone causa subyacente pero esta no ha sido identificada, tras el estudio etiológico, son denominados criptogénicos y representan el 20% restante. El grupo criptogénico presenta al inicio de los espasmos un desarrollo psicomotor normal, que se ve deteriorado progresivamente en la mayoría de los casos. Si bien, una pequeña parte de ellos, aproximadamente el 5% de los espasmos infantiles, presenta una evolución favorable con resolución completa de los espasmos y desarrollo psicomotor normal, lo que ha llevado a algunos autores a considerarlo un grupo etiológico aparte denominado idiopático5, aunque este no ha sido reconocido hasta el momento por la ILAE. Esta clasificación etiológica se basa en la evolución del cuadro, por lo que solo puede establecerse mediante el seguimiento evolutivo de los pacientes.
El deterioro en el desarrollo psicomotor, presente en los pacientes al diagnóstico, puede ser explicado tanto por la enfermedad subyacente como por la propia encefalopatía epiléptica (espasmos no identificados con anterioridad o presencia de hipsarritmia previa al comienzo clínico), por lo que algunos autores proponen abandonar la distinción entre sintomático y criptogénico en función del desarrollo neurológico previo. De acuerdo al United Kingdom Infantile Spasms Study (estudio UKISS) el 61% de los pacientes presentaba una etiología claramente probada, el 33% no tenía causa identificada y el 6% tuvo un estudio incompleto4. Un estudio reciente de la ILAE propone 3 nuevos grupos etiológicos: genético, estructural-metabólico y desconocido6.
A la hora de establecer el diagnóstico etiológico del SW la neuroimagen es la prueba con mayor rentabilidad diagnóstica. En aproximadamente el 70% de los pacientes se alcanza la causa subyacente tras una correcta anamnesis, examen físico y la realización de una resonancia magnética craneal7. Varios estudios evalúan el rendimiento de otras pruebas diagnósticas como son los estudios genéticos y el análisis metabólicos6,8,9. Indican, en aquellos niños sin diagnóstico etiológico tras el estudio inicial, que un estudio coste-eficiente sería la realización de un CGHa, seguido de un panel de epilepsia y un estudio metabólico general (ácido láctico, ácido pirúvico, aminoácidos y ácidos orgánicos en orina) en aquellos en los que el CGHa no fuese definitivo. De este modo se alcanzaría el diagnóstico etiológico en un 10-15% adicional de casos. El estudio realizado por el National Infantile Spasms Consortium de los EE. UU. concluyó que la realización de un CGHa arrojó el diagnóstico en un 11%, el panel de epilepsia en un 31% y el estudio metabólico en un 4,5% de los pacientes sin diagnóstico etiológico previo.
El avance en las técnicas diagnósticas, así como el interés en la causa subyacente debido a la información pronóstica que genera, ha propiciado la reducción del número de casos criptogénicos a favor de los sintomáticos.
Los espasmos infantiles son resistente a la mayoría de los fármacos antiepilépticos; solo 2 terapias se han mostrado claramente eficaces en el SW, la vigabatrina (VGB) y la hormona adrenocorticotropa (ACTH). A día de hoy, como bien indica la última revisión de la Cochrane, no disponemos de protocolos que aboguen a favor de uno sobre otro como tratamiento inicial. En algunos estudios se ha demostrado que el tratamiento con ACTH produce un mayor efecto sobre el control de los espasmos a corto plazo, aunque no se ha podido demostrar su superioridad a largo plazo10–12.
Disponemos de numerosos estudios y revisiones que tratan sobre el SW de forma global, mostrando especial énfasis en la forma sintomática debido a su mayor prevalencia. En cambio, son pocos los que analizan de forma específica el subgrupo criptogénico, siendo este por tanto menos conocido y de pronóstico variable.
El objetivo de este estudio es definir el perfil del SW criptogénico en nuestro medio, atendiendo a sus características clínicas, estudios diagnósticos realizados, tratamiento recibido y evolución posterior. En base a ello, establecemos factores pronósticos que ayuden a mejorar su desarrollo posterior y al asesoramiento de las familias.
Pacientes y métodoSe realizó un estudio observacional retrospectivo mediante revisión de historia clínica digitalizada en 16 pacientes. El análisis estadístico empleado fue únicamente de tipo descriptivo, sin recurrir a métodos analíticos. Los criterios de inclusión fueron: etiología criptogénica (entendida como desarrollo psicomotor normal previo al diagnóstico), exploración neurológica al diagnóstico sin signos de focalidad, ausencia de crisis previas a los espasmos (excluidas crisis neonatales y febriles), neuroimagen normal y no presentar causa etiológica atribuible; diagnóstico establecido en el período 2000-2015; y tratamiento y seguimiento en nuestro centro. El tiempo medio de seguimiento fue 6,6 años, con un tiempo mínimo de 2 años y máximo de 15 años. No se produjo ninguna pérdida durante el seguimiento.
Se elaboró una hoja de recogida de datos que incluía las siguientes variables: sexo, edad de inicio, antecedentes (obstétricos, personales y familiares), desarrollo psicomotor al inicio, crisis epilépticas antes de la aparición de los espasmos, tipo de espasmos, anomalía en el EEG, tiempo de demora en el inicio del tratamiento, tratamiento inicial, respuesta al tratamiento, persistencia de espasmos y/o hipsarritmia, tratamientos recibidos, efectos adversos, recaída, desarrollo psicomotor durante la evolución, aparición de otras epilepsias, tiempo libre de crisis y desarrollo de enfermedad neurológica.
De acuerdo con el esquema terapéutico empleado en nuestro servicio, todos los pacientes recibieron como tratamiento de primera línea VGB en dosis ascendentes (hasta 200mg/kg/día). Si el tratamiento en monoterapia resultó ineficaz tras un plazo de 2 semanas (persistencia de espasmos y/o hipsarritmia), se asoció tratamiento con ACTH con dosis diaria durante 14 días y descenso paulatino posterior en otras 5 semanas.
Entendemos como pronóstico desfavorable el desarrollo posterior de otras formas de epilepsia y/o la aparición de déficit cognitivo moderado/grave según una valoración neuropsicológica.
ResultadosSe estudiaron un total de 16 pacientes, sin producirse ninguna pérdida durante el seguimiento evolutivo.
En cuanto al sexo encontramos un predominio de varones, con 11/16 (69%) pacientes de sexo masculino. La edad de presentación osciló entre el mes y medio y los 10 meses de vida, con predominio del grupo entre 5 y 7 meses (50%). La edad media al inicio de los espasmos fue de 6 meses.
Respecto a los antecedentes; 6/16 presentaban antecedente familiar de algún tipo de epilepsia, todas ellas epilepsias benignas idiopáticas en familiares de primer o segundo grado; y 2/16 tenían antecedente patológico de crisis previa a la aparición de los espasmos, uno en forma de crisis neonatal y otro como crisis febril.
Los espasmos más frecuentes fueron los de tipo extensor (50%), seguidos de los flexores (43%) y los mixtos (7%). Solo 4/16 pacientes presentaban un retraso psicomotor leve al inicio de los espasmos.
El tiempo transcurrido entre la aparición de los espasmos y el inicio del tratamiento fue inferior a una semana en el 50% de los pacientes. De acuerdo al esquema terapéutico del servicio, todos los pacientes recibieron como tratamiento de inicio VGB (fig. 1).

La respuesta a la VGB fue favorable en 5/16, todos ellos presentaron desaparición de los espasmos durante la primera semana de tratamiento y normalización del EEG en 2 semanas. Aquellos pacientes que no presentaron mejoría tras 15 días de tratamiento con VGB recibieron como tratamiento de segunda línea ACTH. De los 11 pacientes tratados con ACTH, se produjo una respuesta favorable con control de la hipsarritmia en la totalidad de ellos.
Se presentaron efectos secundarios en el 32% de los pacientes, la mayoría de ellos asociado al tratamiento con ACTH. De los pacientes tratados con VGB solo 1/16 presentó reacción adversa en forma de estado encefalopático que mejoró a las pocas horas de su retirada. De los tratados con ACTH, 4/11 presentaron hipertensión arterial que requirió tratamiento hipotensor; 2 de ellos asociaron además cambios ecocardiográficos en forma de hipertrofia septal, esta resultó transitoria y mejoró tras finalizar el tratamiento con ACTH.
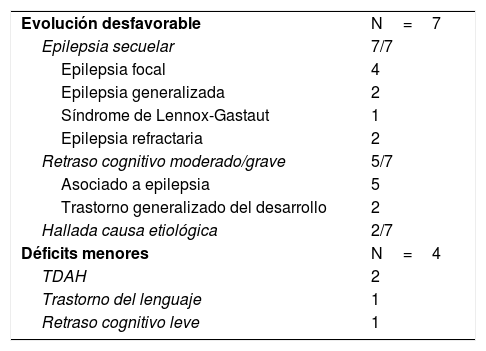
El pronóstico fue desfavorable en 7/16 pacientes (43,7%), con evolución hacia otras formas de epilepsia secuelar en estos 7 casos y retraso cognitivo moderado/grave en 5 de ellos, 2 de ellos en el contexto de un trastorno generalizado del desarrollo. El tiempo medio que permanecieron libres de crisis hasta el desarrollo de otra forma de epilepsia fue de 10 meses, apareciendo en todos ellos en los primeros 2 años del diagnóstico. Otros pacientes presentaron déficits menores a largo plazo: trastorno del lenguaje, déficit de atención e hiperactividad y retraso cognitivo leve, si bien aunque se recogen en los resultados, estos no se incluyeron en el grupo con pronóstico desfavorable al no mostrar repercusión funcional importante en su vida diaria. No se produjeron recaídas (tabla 1).
Evolución
| Evolución desfavorable | N=7 |
| Epilepsia secuelar | 7/7 |
| Epilepsia focal | 4 |
| Epilepsia generalizada | 2 |
| Síndrome de Lennox-Gastaut | 1 |
| Epilepsia refractaria | 2 |
| Retraso cognitivo moderado/grave | 5/7 |
| Asociado a epilepsia | 5 |
| Trastorno generalizado del desarrollo | 2 |
| Hallada causa etiológica | 2/7 |
| Déficits menores | N=4 |
| TDAH | 2 |
| Trastorno del lenguaje | 1 |
| Retraso cognitivo leve | 1 |
N: tamaño muestral; TDAH: trastorno por déficit de atención e hiperactividad.
Durante el seguimiento, en 2 de los 16 pacientes se llegó finalmente a la causa etiológica. Un déficit de biotinidasa tras la ampliación del estudio metabólico en uno de los pacientes que presentaba una dermatitis de tórpida evolución, y una displasia cortical focal evidenciada a los 10 años del diagnóstico; ambos pertenecientes al subgrupo con pronóstico desfavorable. Por tanto, el pronóstico global de nuestros pacientes criptogénicos mejoraría al excluir de los resultados a estos 2 pacientes (que pasaron a ser sintomáticos), presentando una evolución desfavorable solo 5/14 pacientes (un 35,7%).
Se estudiaron distintas variables mediante análisis descriptivo, en busca de posibles factores que interfieran en el pronóstico, y así plantear hipótesis de trabajo para futuros estudios analíticos. Se encontró mayor evolución hacia otras formas de epilepsia en el sexo masculino (55%) y en el inicio antes de los 6 meses (71%), en cambio, el tratamiento temprano (en las primeras 2 semanas del comienzo de los espasmos) y el control precoz de la hipsarritmia (en las primeras 3 semanas) tuvieron una menor evolución hacia epilepsia secuelar (36 y 33% respectivamente) (tabla 2).
Posibles factores pronósticos
| Factor | Epilepsia secuelar | |
|---|---|---|
| Sexo masculino (n=11) | 55% | 6/11 |
| Sexo femenino (n=5) | 20% | 1/5 |
| Comienzo antes de los 6 meses de edad (n=7) | 71% | 5/7 |
| Comienzo pasados los 6 meses de edad (n=9) | 22% | 2/9 |
| Con AF de epilepsia (n=6) | 33% | 2/6 |
| Sin AF de epilepsia (n=10) | 50% | 5/10 |
| Tratamiento con VGB en monoterapia (n=5) | 40% | 2/5 |
| Tratamiento con VGB y ACTH (n=11) | 45% | 5/11 |
| Demora tratamiento mayor de 2 semanas (n=5) | 60% | 3/5 |
| Demora tratamiento menor de 2 semanas (n=11) | 36% | 4/11 |
| Persistencia de hipsarritmia mayor de 3 semanas (n=4) | 75% | 3/4 |
| Persistencia de hipsarritmia menor de 3 semanas (n=11) | 33% | 4/11 |
ACTH: hormona adrenocorticotropa; AF: antecedentes familiares; n: tamaño muestral; VGB: vigabatrina.
No hemos encontrado diferencias, respecto al resto de trabajos publicados, en cuanto a edad de comienzo de los espasmos, predominio del sexo masculino y tipo de espasmos.
Analizando en nuestro estudio de forma específica el subgrupo criptogénico, sí encontramos una mayor presencia de antecedentes familiares de epilepsia. En estudios previos referidos al SW de manera global, la historia familiar de epilepsia oscila entre el 15-25%1,3, ascendiendo en nuestra serie de casos criptogénicos al 37%. Este hecho estaría en consonancia con la importancia de los factores genéticos en este grupo de pacientes, algo ya indicado en otras revisiones que relacionan el SW sin etiología reconocida con otras formas idiopáticas de epilepsia, como es la epilepsia de ausencia juvenil5.
Varios estudios han encontrado una clara relación entre el inicio precoz del tratamiento y una posterior evolución favorable, principalmente en los de tipo criptogénico9,13–17. En este estudio, el 50% de los pacientes fueron remitidos a nuestro centro precozmente, iniciándose el tratamiento en la primera semana de aparición de los espasmos. En 5 pacientes la demora en el tratamiento fue superior a las 2 semanas, evolucionando de manera desfavorable 3 de ellos (60%) con desarrollo posterior de otras formas de epilepsia, lo que supone también una peor evolución respecto al pronóstico global. En cambio, de los 11 pacientes que iniciaron tratamiento en las primeras 2 semanas del comienzo, solo 4 (36%) evolucionaron a epilepsia secuelar.
Desde que Chiron et al.18 demostraran la eficacia de la VGB en el control de los espasmos, numerosos autores han comparado esta con el tratamiento con ACTH, evidenciando una respuesta similar a largo plazo aunque con menor tasa de efectos adversos11,12,19. El esquema terapéutico de nuestro servicio, basado en el estudio europeo de Aicardi20–22, emplea como ya se ha mencionado tratamiento de primera línea con VGB en monoterapia, reservando la terapia con ACTH para aquellos en los que fracasó el tratamiento de inicio.
En nuestro estudio se consideró como respuesta inicial favorable la desaparición de los espasmos y normalización del EEG durante las 2 primeras semanas de tratamiento, siempre que esto persistiera al menos durante un mes. El tratamiento de inicio con VGB tuvo una respuesta inicial favorable en el 30% de los casos criptogénicos recogidos, porcentaje menor al 50-70% obtenido en otras series1,19,23. De estos pacientes, con respuesta inicial favorable a la VGB en monoterapia, el 60% se mantuvo libre de crisis durante el seguimiento posterior, porcentaje muy similar al encontrado en la evolución a largo plazo de los pacientes tratados con VGB y ACTH. Por tanto, aunque la respuesta inicial a la VGB fuera menor a la esperada, el pronóstico a largo plazo independientemente del tratamiento recibido fue favorable en el 56% de los casos, resultado similar al de otros autores9,10,14 (fig. 2).

La retinopatía gabaérgica, causante de defectos campimétricos periféricos irreversibles en el adulto, sigue siendo a día de hoy el efecto adverso más significativo del tratamiento con VGB, si bien en edad pediátrica resulta difícil de identificar. Distintos estudios lo relacionan directamente con la dosis y el tiempo de tratamiento, considerando seguro el empleo por período inferior a 6 meses y en dosis que no sobrepase 200mg/kg/día15,24,25. De los 16 pacientes seguidos, ninguno de ellos presentó dicha complicación, pese a prolongarse el tratamiento durante más tiempo en la mayoría de ellos, con máximo de 3 años. En cambio, el tratamiento con ACTH produjo efectos adversos moderados que precisaron tratamiento en el 36% de los casos.
El control de la toxicidad retiniana resultó difícil de valorar tanto por la edad pediátrica como por la discapacidad intelectual propia de alguno de estos pacientes, si bien las familias no refirieron problemas visuales en el día a día en ninguna de las consultas de seguimiento. Todos realizaron revisiones periódicas por Oftalmología con análisis grosero del campo visual (reflejo de amenaza, aproximación de objetos y campimetría por confrontación) sin indicar déficits. El estudio mediante campimetría computarizada solo se realizó en 2 pacientes, ambos sin discapacidad intelectual y una vez alcanzada la edad prepúber, con resultados en ambos casos dentro de la normalidad, lo que nos lleva a pensar que no presentaron afectación retiniana o bien esta fue reversible.
Por este motivo, en contra de lo indicado por otros autores que abogan por el tratamiento de primera línea con ACTH26, no encontramos un peor pronóstico a largo plazo en los tratados únicamente con VGB y su perfil de seguridad resultó favorable en esta serie.
El pronóstico del SW de etiología criptogénica continúa siendo grave. El 44% de los pacientes desarrollaron otra forma de epilepsia (en el 71% de ellos asociada a un déficit cognitivo moderado-grave), datos bastante próximos a los de otras publicaciones9,10,14. La incidencia del síndrome de Lennox-Gastaut fue baja (1/16), siendo la epilepsia focal la evolución más frecuente. El paciente que desarrolló síndrome de Lennox-Gastaut no respondió a VGB en monoterapia, precisando asociación de ACTH; pese a ello el tiempo trascurrido hasta la desaparición de la hipsarritmia fue de 3 meses y medio desde el inicio del tratamiento. Por ello, aunque se inició el tratamiento durante la primera semana de aparición de los espasmos, la persistencia de la hipsarritmia sobre un cerebro inmaduro podría haber contribuido a la peor evolución14,27,28.
Al incluir en nuestro estudio solo a pacientes de etiología criptogénica, cabría esperar una mejor evolución a largo plazo. En cambio, el pronóstico favorable apenas alcanza el 55%, probablemente debido a la presencia de causa orgánica no identificada en algunos de ellos, causas que ensombrecerían al pronóstico per se independientemente del control de la hipsarritmia. El avance en las nuevas técnicas diagnósticas, así como el mayor empleo de análisis genéticos, identificará progresivamente más causas que justifiquen esta encefalopatía epiléptica, seleccionando cada vez más a los pacientes criptogénicos y mejorando por tanto su pronóstico a largo plazo6,29,30. En nuestro caso, debido a la falta de disponibilidad en nuestro centro durante el período referido, el estudio genético se limitó a la realización de cariotipo y detección de síndromes microdelecionales, sin poder acceder a la solicitud de paneles genéticos. Si bien, mediante estudio metabólico y de neuroimagen, se identificó finalmente una causa subyacente en 2/16 pacientes, que si son excluidos de los resultados el pronóstico favorable ascendería al 64% aproximándose a lo esperado inicialmente.
Reconocemos las limitaciones de nuestro estudio, lo que nos lleva a defender futuras investigaciones en este campo. Aspectos como un tamaño muestral reducido, el empleo de un análisis puramente descriptivo, limitación en el acceso a técnicas diagnósticas que delimiten el grupo criptogénico y la subjetividad a la hora de valorar la afectación retiniana serían aspectos a mejorar.
En conclusión, pese a que nuestra serie se limita a casos catalogados inicialmente como criptogénicos, encontramos un porcentaje importante con pronóstico desfavorable a largo plazo, lo que reafirma la gravedad de este cuadro. El estudio etiológico exhaustivo en los pacientes con SW continúa siendo imprescindible, con el objeto de delimitar adecuadamente el subgrupo criptogénico y poder conocer su pronóstico real. Entre los posibles factores estudiados ligados al buen pronóstico encontramos el sexo femenino, el comienzo de los espasmos pasados los 6 meses, la instauración temprana del tratamiento y el control precoz de la hipsarritmia, cobrando un especial interés estos 2 últimos al ser elementos sobre los que podemos influir. En cambio, el tratamiento de primera línea con VGB en monoterapia no se asoció a un mayor desarrollo de epilepsia secuelar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este artículo fue presentado parcialmente en formato póster en la XXXIX reunión de la SENEP el pasado 19-21 de mayo de 2016 en Toledo.
