









La enfermedad de células falciformes (ECF), pese a la mejora en el manejo médico, persiste asociada a morbilidad y a menor supervivencia. El alotrasplante de progenitores hematopoyéticos (alo-TPH) es actualmente la única opción curativa. Describir la evolución clínico-analítica de los pacientes trasplantados en nuestro centro.
Material y métodoEstudio unicéntrico descriptivo, incluye a pacientes con ECF en los que se realiza alo-TPH de médula ósea de hermano HLA-idéntico desde enero del 2010 hasta diciembre del 2014. Se recogen datos epidemiológicos, clínicos y analíticos con tiempo de seguimiento hasta diciembre del 2015. Los datos se presentan como frecuencias, porcentajes y medianas (rango).
ResultadosSe recluta a 11 pacientes (8 varones), mediana de edad: 7 años (2-13), todos ellos con comorbilidad previa al TPH. Se consigue injerto estable en 10/11 pacientes, quimerismo completo en 9/11 y quimerismo mixto estable tras un año del TPH en 1/11. Un paciente presenta fallo secundario de injerto con reaparición de clínica el día +180. Complicaciones post-TPH: complicaciones neurológicas 4/11 pacientes (hemorragia subaracnoidea, crisis), HTA 7/11, fallo renal agudo 3/11, reactivación CMV 9/11, EICHa cutáneo 6/11, uno de ellos desarrolla EICH intestinal grado iv causando su fallecimiento (día +51). Ningún paciente desarrolla EICH crónico. Supervivencia global y libre de eventos a los 3,1 años de seguimiento: 90,9 y 81,9%, respectivamente.
ConclusionesEl alo-TPH, única opción curativa, no está exento de morbimortalidad, encontramos un riesgo de muerte similar a otras series (1/11), siendo su primera causa el EICH agudo. Otros problemas son fallo de injerto (1/11) y complicaciones neurológicas (4/11), aunque las secuelas permanentes son leves.
Sickle cell disease (SCD), despite the improvement in the medical management, is still associated with severe morbidity and decreased survival. Allogenic hematopoietic stem cell transplantation (Allo-HSCT) currently provides the only curative therapy. A report is presented on our experience in children with SCD, who underwent Allo-HSCT in a single centre.
Material and methodA single centre descriptive study was conducted on patients with SCD who underwent a bone marrow transplant from an HLA-identical sibling donor between January 2010 and December 2014. Epidemiological, clinical and analytical parameters were collected with a follow-up to December 2015. Data are presented as frequencies, percentages, and medians (range).
ResultsAllo-HCST was performed in 11 patients (8 males) with a median age of 7 years (2-13), all of them with comorbidity prior to the HCST. A stable graft was achieved in 10 out of 11 patients, 9 of them with complete donor chimerism, and one patient with stable mixed chimerism after 1 year of allo-HSCT. One patient has secondary graft failure with re-appearance of symptoms associated with SCD on day 180. Complications of Allo-HSCT are: arterial hypertension 7/11, acute renal failure 3/11, CMV reactivation 9/11, neurological complications 4/11 (subarachnoid haemorrhage, seizure), and acute graft versus host disease (aGVHD) of the skin 6/11, one of whom developed grade iv intestinal aGVHD, causing his death (day 51). None of the patients developed chronic GVHD. The overall survival and event-free survival was 90.9% and 81.9%, respectively, with a median follow-up of 3.1 (1-5.7) years.
ConclusionsAllo-HSCT, the only curative therapy, remains associated with morbidity. There was a transplant related mortality in our study, consistent with multicentre studies (1/11), and with aGVHD being the main cause. Other problems still include graft failure (1/11), and neurological complications (4/11), although the permanent sequelae are mild.
La enfermedad de células falciformes (ECF) es actualmente la alteración más prevalente identificada en los estudios de cribado neonatal de varios países, entre ellos España. En la Comunidad de Madrid, donde se realiza el cribado desde mayo del 2003, se confirma este dato con una incidencia de 1 por cada 5.000 recién nacidos vivos1. La ECF es causa de importante morbilidad multisistémica y de un aumento del riesgo de muerte precoz2. En las últimas 3 décadas, diversas medidas, como la introducción de penicilina profiláctica, la vacunación frente al Streptococcus pneumoniae, el uso de hidroxiurea o el régimen hipertransfusional han disminuido la morbimortalidad en los pacientes afectados de ECF3-5. La hidroxiurea ha demostrado reducir de forma significativa la frecuencia de crisis vaso-oclusivas y de síndromes torácicos agudos6,7; sin embargo, no revierte el daño producido en los órganos y no disminuye el riesgo de accidente cerebrovascular (ACV). La introducción de la ecografía Doppler transcraneal (EDTC) en el seguimiento permite detectar aquellos con alto riesgo de desarrollar ACV agudos y el uso de la resonancia magnética (RM) cerebral el diagnóstico de los infartos silentes8. En la prevención primaria de ACV, el régimen hipertransfusional ha demostrado su utilidad, pero implica la sobrecarga férrica secundaria y un incumplimiento a largo plazo9.
El trasplante de progenitores hematopoyéticos (TPH) es actualmente la única opción curativa. El primer caso de TPH con éxito se realizó en 1984 en Estados Unidos en un paciente con drepanocitosis y LMA7. Estudios multicéntricos posteriores han obtenido buenos resultados con una supervivencia global del 93-94% y libre de eventos del 82-86%, siendo las principales complicaciones el fallo de injerto y la enfermedad injerto contra huésped (EICH)10-14.
El objetivo de nuestro estudio es describir la evolución clínica y analítica de los pacientes con ECF trasplantados de un hermano HLA-idéntico en nuestro centro.
Materiales y métodosEstudio unicéntrico descriptivo retrospectivo. Se recoge a aquellos pacientes pediátricos afectados de ECF con forma homocigota (HbSS) o doble heterocigota (HbSβ0), que reciben un trasplante de médula ósea de un hermano HLA idéntico (fenotipo de Hb AA o Hb AS) desde enero de l2010 hasta diciembre del 2014, con un tiempo de seguimiento hasta diciembre del 2015. Las variables estudiadas son demográficas (edad, sexo) y antecedentes del paciente relacionados con complicaciones propias de la enfermedad: síndrome torácico agudo (STA), crisis vaso-oclusiva (CVO), dactilitis, priapismo, ACV, alteración en la ecografía Doppler trascraneal, necrosis avascular de cadera, secuestro esplénico y necesidad de esplenectomía. En relación con el tratamiento de base previo al TPH, se señala si reciben hidroxiurea (HU), régimen hipertransfusional (RHT) y la necesidad de quelación del hierro. Otras variables estudiadas en relación con el TPH incluyen compatibilidad ABO entre donante y receptor, celularidad infundida, día de injerto de plaquetas y neutrófilos así como la evolución en el quimerismo tanto en médula ósea en el día +30, como posteriormente en sangre periférica. Los parámetros analíticos seleccionados de forma basal, a los 3, 6, 9 y 12 meses del TPH son: hemoglobina, reticulocitos, LDH, bilirrubina, ferritina, filtrado glomerular por cistatina C, subpoblaciones linfocitarias (linfocitos B, T, NK), inmunoglobulinas (Ig). Por último, se incluyen las complicaciones post-TPH en cuanto a EICH aguda (EICHa), complicaciones neurológicas (crisis, hemorragia subaracnoidea [HSA], neurotoxicidad), insuficiencia renal aguda, hipertensión arterial (HTA), reactivación del CMV y causa del fallecimiento.
Régimen de acondicionamientoEl acondicionamiento es mieloablativo, consistente en busulfán iv (estándar en función del peso, entre 3,2 y 4,8mg/kg/día, en dosis única cada 24h, los días –9 al –6), alemtuzumab iv (0,1 mg/Kg/día, los días –8 al –6) y ciclofosfamida iv (50mg/kg/día, los días –5 al –2, junto con Mesna). La profilaxis de EICH se realiza desde el día –1 con ciclosporina iv durante 3-6 meses y 2 dosis de metotrexato iv (días +4 y +7). Se monitoriza la PCR de CMV, VEB y serología de Aspergillus, y profilaxis infecciosa con micafungina, levofloxacino y aciclovir, así como cotrimoxazol tras el prendimiento. Otros cuidados incluyen la administración de Ig, ursodesoxicólico, fenitoína, antihipertensivos para mantener PA alrededor del percentil 95, aportes de magnesio si se precisa y soporte transfusional empleando productos irradiados. El procedimiento se lleva a cabo en habitaciones individuales con sistemas de aire con filtros HEPA y presión positiva.
Todos los pacientes reciben transfusiones mensuales o exanguinotransfusión para obtener Hb S < 30% previa al TPH y durante el mismo se mantienen plaquetas>50.000/μL y Hb entre 11-13g/dL. La infusión de progenitores hematopoyéticos es siempre médula ósea en fresco. En el donante del paciente 1 debido a la diferencia de peso con el receptor se realizó extracción de médula ósea en los meses previos y se criopreservó hasta el momento de la infusión (día +1), 24 h después de infundir la médula ósea en fresco.
Estudio estadísticoEn el análisis de datos se utilizó el programa SPSS®. Los datos se presentan como frecuencias, porcentajes, medias y medianas (rangos). En la comparación de variables cuantitativas apareadas se emplea el test de Wilcoxon, considerándose estadísticamente significativos valores de p<0,05.
Estudio de supervivencia global y libre de eventos utilizando la curva de Kaplan y Meier. Los eventos se definen como muerte, pérdida de injerto (quimerismo con > 95% del receptor) o reaparición de la clínica de ECF.
DefinicionesSe definen como día de injerto de neutrófilos y plaquetas, el primero de 3 días consecutivos con recuento en número absolutos de>500 neutrófilos/μL o > 50.000 plaquetas/μL. Se define quimerismo completo cuando > 95% de las células hematopoyéticas proceden del donante.
Se define fallo de injerto primario cuando en el día +28 post-TPH no se alcanzan en sangre periféricas los valores de neutrófilos>500/μL, plaquetas>50.000/μL, Hb>8g/dL y fallo de injerto secundario la pérdida de al menos 2 líneas celulares previamente implantadas y funcionales. Se refine rechazo o reconstitución autóloga cuando reaparecen síntomas de la ECF.
ResultadosCaracterísticas de los pacientes (tabla 1)Desde enero del 2010 hasta diciembre del 2014, 11 pacientes (8 varones y 3 mujeres) reciben alo-TPH de médula ósea de su hermano HLA-idéntico. Todos ellos tienen un fenotipo de hemoglobina S homocigota (Hb SS). La mediana de edad es 7 años (2-13). En relación con la morbilidad previa al TPH, presentan CVO recurrente (n=9), STA recurrente (n=6), ACV (n=2), velocidad alterada en la EDTC (n=4), esplenectomía (n=2), atrofia testicular (n=1). El tratamiento basal previo al TPH es hidroxiurea en 4 pacientes, uno régimen hipertransfusional y 3 tanto hidroxiurea como transfusiones periódicas. Dos pacientes presentan sobrecarga férrica precisando quelación de hierro.
Características de los pacientes y sus donantes
| N.° paciente | Sexo | Edad al TPH | Tto. hidroxiurea | Indicación TPH | Comorbilidades | Donante | ||
|---|---|---|---|---|---|---|---|---|
| Tipo Hb | ABO Receptor/donante | IgG CMV Receptor/donante | ||||||
| Pac. 1 | V | 13 a, 10m | Sí | EDTC alterada, CVO, STA | Hb AA | A Rh+/O Rh+ | +/+ | |
| Pac. 2 | M | 7 a, 10m | No | ACV, EDTC alterada | Sd. Moya-Moya | Hb AA | AB Rh+/AB Rh- | +/+ |
| Pac. 3 | M | 10 a, 5m | Sí | EDTC alterada, CVO | Hb AA | A Rh+/B Rh+ | +/+ | |
| Pac. 4 | V | 5 a, 6m | Sí | CVO, STA | Atrofia teste dcho. Teste izdo. con microcalcificac. | Hb AA | A Rh+/A Rh + | +/+ |
| Pac. 5 | V | 9 a, 8m | Sí | ACV, EDTC alterada, CVO | Hb AS | B Rh+/O Rh+ | +/+ | |
| Pac. 6 | M | 4 a, 9m | No | STA, CVO | Hb AS | B Rh+/B Rh+ | +/+ | |
| Pac. 7 | V | 9 a, 2m | Sí | STA, CVO | Hb AS | O Rh+/O Rh+ | +/+ | |
| Pac. 8 | V | 2 a, 7m | No | STA, CVO | Hb AA | A Rh+/A Rh+ | –/+ | |
| Pac. 9 | V | 9 a, 10m | Sí | STA | Esplenectomizado Foco de no compactación en ventrículo izdo. | Hb AS | O Rh+/B Rh– | +/– |
| Pac. 10 | V | 2 a, 1m | No | CVO | Hb AS | A Rh+/O Rh+ | +/+ | |
| Pac. 11 | V | 6 a, 1m. | Si | CVO | Esplenectomizado | Hb AS | O Rh+/O Rh+ | –/+ |
Todos los donantes de médula ósea son hermanos HLA idénticos, el 55% de ellos con fenotipo de rasgo falciforme (Hb AS). Existe incompatibilidad menor de grupo en 4 pacientes y mayor en 2.
Injerto y complicaciones post-trasplante de progenitores hematopoyéticos (tabla 2)Se consigue injerto estable en 10 de los 11 pacientes, 9 de ellos con quimerismo completo.
Trasplante de progenitores hematopoyéticos (TPH), resultados y complicaciones post-TPH
| N.° Pac. | Celularidad infundida: CD34×10e6/Kg | Día injerto Nts | Día injerto Plaqs. | Quimerismo (% del receptor) | Reactivación CMV (día y tratamiento) | |
|---|---|---|---|---|---|---|
| Día+30 (MO) | Última quimera (tiempo post-TPH) | |||||
| Pac. 1 | Día 0 (MO fresco): 3,8 Día +1 (MO descongelada): 6.1 | +16 | +31 | 1,9% | 4,4% (5 a, 7m) | Sí, +9, tto. foscarnet |
| Pac. 2 | 6 | +23 | +27 | 0% | 4,8% (4 a, 8m) | Si, +13, tto. foscarnet |
| Pac. 3 | 3,4 | +23 | +27 | 0% | 3,6% (4 a, 7m) | Sí, +11, tto. foscarnet |
| Pac. 4 | 8,5 | +27 | +27 | 0% | 3,6% (4 a, 1m) | No |
| Pac. 5 | 5,48 | +16 | +38 | 0% | 0% (3 a, 1m) | S, +13, tto. foscarnet |
| Pac. 6 | 4,63 | +18 | +42 | 0% | 0% (3 a, 9m) | Sí, +7, tto. foscarnet |
| Pac. 7 | 3,72 | +20 | +19 | 0% | Sí, +39, tto. ganciclovir | |
| Pac. 8 | 6,52 | +25 | +39 | 0% | 0% (1 a, 9m) | Sí, +47, tto. ganciclovir |
| Pac. 9 | 2,85 | +18 | +28 | 1,3% 51% Lin T | 92% 88% Lin T (1 a, 7m) | Sí, +8, tto. foscarnet |
| Pac. 10 | 5,3 | +24 | +94 | 2,4% | 0% (1 a, 2m) | Sí, +9, tto. foscarnet |
| Pac. 11 | 3,5 | +22 | +24 | 2,5% 59% Lin T | 6% 12% Lin T (1 a, 0m) | No |
| N.° Pac. | Complicaciones neurológicas agudas (día de presentación) | HTA (día inicio) | IRA | EICHa-localización y grado | Otras complicaciones post-TPH | Evento (fallecido/pérdida injerto/reaparición clínica ECF/vivo) | |
|---|---|---|---|---|---|---|---|
| Crisis | HSA | ||||||
| Pac. 1 | Sí, +1 +27 | Sí, +1 | Sí (+1) | No | Sí Cutáneo II | Día +27: RM cerebral: toxicidad por ciclosporina Déficit cognitivo leve Mucositis grado 3 | Vivo |
| Pac. 2 | No | No | Si (día 0) | No | No | Vivo | |
| Pac. 3 | Sí, +29 | No | Sí (+17) | No | Sí Cutáneo I | Vivo | |
| Pac. 4 | No | No | No | No | No | Mucositis grado 4 | Vivo |
| Pac. 5 | No | No | Sí (+13) | Sí | Sí Cutáneo II | Infección posible por Aspergillus | Vivo |
| Pac. 6 | No | No | No | No | No | Infección relacionada con catéter y miosistis de glúteo izdo. por SAMR | Vivo |
| Pac. 7 | Sí, +1 | Sí, +1 | Sí (+1) | Sí | Sí Cutáneo IV Digestivo IV Hepático III | Cistitis hemorrágica por virus BK Colestasis y desarrollo de barro biliar y colelitiasis Fiebre sin aislamiento microb. | Fallecido día +51 (EICHa grado iv y fallo multiorgánico) |
| Pac. 8 | No | No | No | Ni | Sí Cutáneo I | Mucositis grado 4 | Vivo |
| Pac. 9 | No | Sí, +2 | Sí (día –1) | No | No | Mucositis grado 1 Cistitis hemorrágica por virus BK | Reaparición de clínica (CVO) el día +180 |
| Pac. 10 | No | No | Sí (+23) | Sí | Sí Cutáneo III | Mucositis grado 3 Fiebre y neutropenia | Vivo |
| Pac. 11 | No | No | No | No | No | Quimera mixta: se aumenta inmunosupresión por un año | Vivo |
CMV: citomegalovirus; EICHa: enfermedad injerto contra huésped aguda; EICHc: enfermedad injerto contra huésped crónica; HSA: hemorragia subaracnoidea; IRA: insuficiencia renal aguda; Lin T: linfocitos T; MO: médula ósea; Nts: neutrófilos; Plaqs: plaquetas; SP: sangre periférica.
La mediana de injerto de neutrófilos es de 22 días (16-27) y de plaquetas de 28 (19-94).
El paciente 9 (tabla 2) presenta en el día +30 quimerismo completo en médula ósea total (1,3% del receptor), pero el estudio de quimerismo en subpoblaciones celulares permite conocer que el 51% de linfocitos T es del receptor. Se retira la inmunosupresión y se realizan 3 infusiones de linfocitos T del donante en los días +71 (1×105/Kg), +105 (1,95×105/Kg) y +170 (0,85×106/Kg) sin mejoría en la quimera, reapareciendo la clínica de ECF en el día +180 (ingreso por CVO). El paciente 11 (tabla 2) presenta en el día +30 quimerismo completo en médula ósea total (2,5% del receptor) pero mixto en la población de linfocitos T (59% del receptor). Se aumenta la inmunosupresión añadiendo micofenolato a la ciclosporina que se mantienen durante un año. Las cifras de quimerismo mixto permanecen estable: 4,2% a los 3 meses, 6% a los 6 meses, 6% al año con un descenso progresivo del quimerismo del receptor en linfocitos T (12% al año).
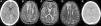
La complicación post-TPH más frecuente es la reactivación del CMV que ocurre en 9 de 11 pacientes, recibiendo tratamiento con foscarnet cuando se produce previo al injerto y valganciclovir si el injerto es estable. La HTA aparece en 7 de 11 niños, en relación con la infusión de progenitores (pacientes 1 y 7), secundario a medicaciones como corticoides para el tratamiento de EICHa (paciente 3), foscarnet por reactivación del CMV (paciente 5), ciclosporina (paciente 9) o bien de causa multifactorial en el contexto de empeoramiento de la función renal (paciente 10). Todos ellos alcanzan buen control posterior de la PA, salvo el paciente 10, que continúa precisando tratamiento por vía oral un año después del TPH, aunque está normotenso con medicación en retirada. En la figura 1 se muestra la evolución del filtrado glomerular por cistatina C en los 3 pacientes que desarrollan insuficiencia renal aguda.
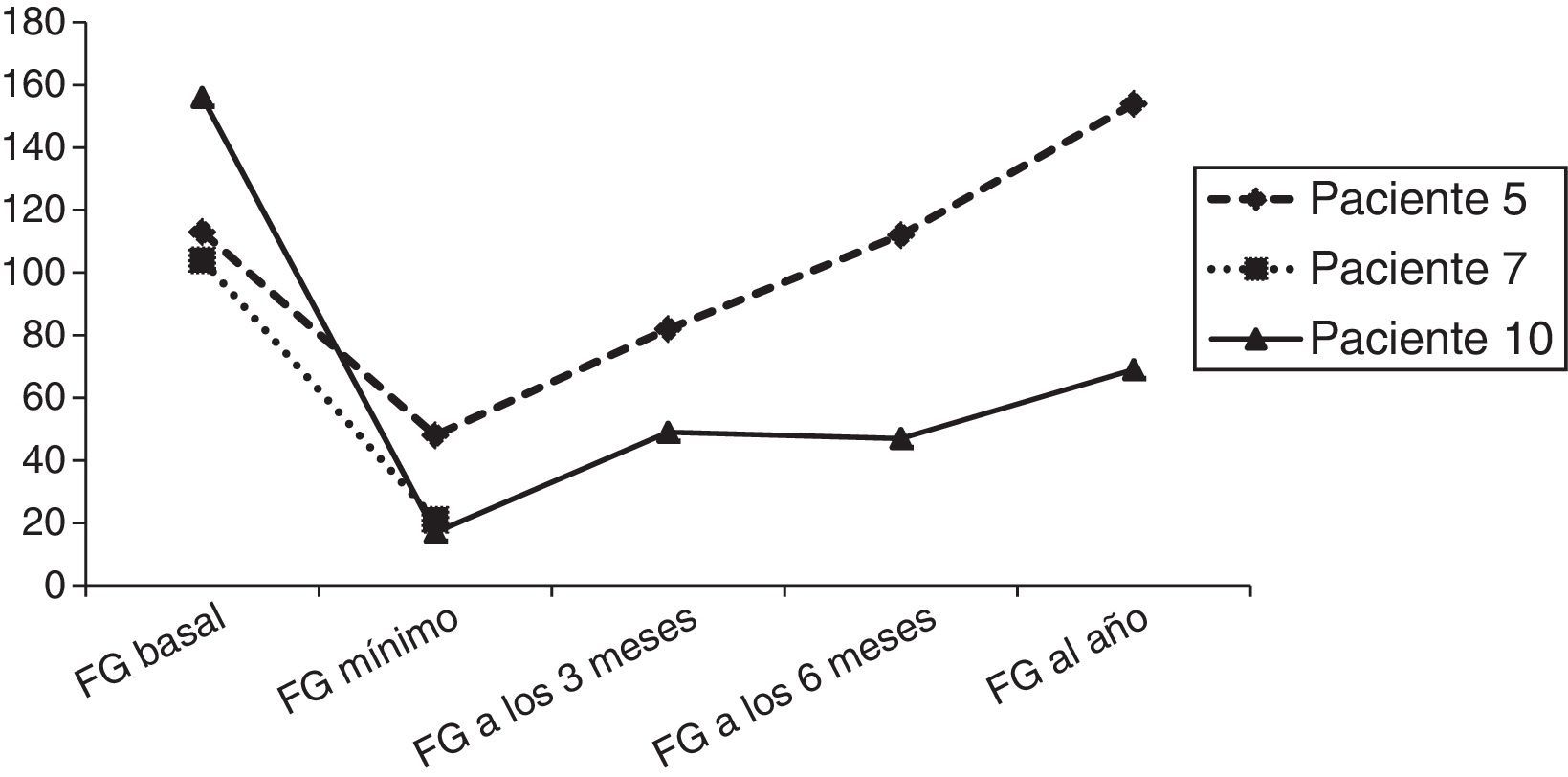
En relación con las complicaciones neurológicas, 3 pacientes presentan HSA en el contexto de HTA, 2 de ellos asociando crisis epilépticas (tabla 2 y fig. 2). El paciente 1 presenta en el día +27 un segundo episodio de crisis, en el TC no se objetivan nuevas zonas de sangrado, aunque aparecen lesiones hipodensas frontales y parieto-occipitales bilaterales. Se amplía estudio con angio-RM cerebral que descarta malformaciones vasculares y objetiva lesiones en ambos hemisferios cerebrales compatibles con toxicidad por ciclosporina y evolución del edema post-HSA. Tras la suspensión de ciclosporina presenta buena evolución neurológica, tanto cognitiva como motora, aunque persiste déficit cognitivo leve. En la RM de control realizada a los 6 meses persisten las lesiones previas en evolución, sin aparición de nuevas (fig. 2). La exploración neurológica de los pacientes con HSA al año del trasplante es normal, el paciente 1ha presentado dificultades escolares (déficit cognitivo leve), sin precisar repetir curso y alcanzando estudios universitarios.
Paciente 1 (A, B, C y D). Imagen de TC de cráneo axial simple donde se evidencia hiperdensidad de los surcos como signo de hemorragia subaracnoidea en la cisura de Silvio del lado derecho, en surcos de la convexidad cerebral, de predominio en el lado derecho y en la cisterna perimesencefálica izquierda (A). Imágenes de RM secuencia TSE T2 axial en el día +27 donde se evidencian lesiones corticosubcorticales frontales bilaterales y lesiones de sustancia blanca profunda frontales bilaterales y parietooccipital izquierda compatibles con toxicidad por ciclosporina (B y C). Imagen de RM TSE T2 axial 6 meses post-TPH donde se evidencian lesiones residuales (D). Paciente 9 (E): imagen de TC de cráneo axial donde se evidencia hiperdensidad de surcos frontotemporales izquierdos como signo de hemorragia subaracnoidea en el día +2.
Seis pacientes desarrollan EICH agudo, 5 de ellos solo con afectación cutánea y buena respuesta a corticoides; el paciente 7 presenta EICHa corticorresistente cutáneo-intestinal grado iv y hepático grado iii, sin respuesta frente a aumento de inmunosupresión (micofenolato, infliximab) y sesiones de fotoaféresis, ocasionando su fallecimiento en el día +51. Ningún paciente desarrolla EICH crónico ni enfermedad veno-oclusiva hepática.
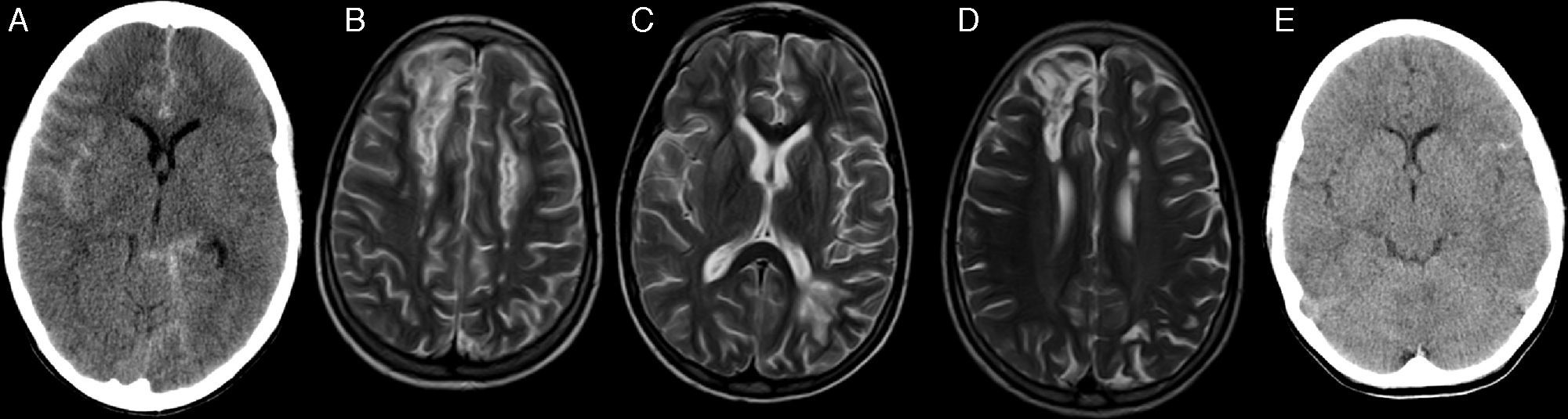
Actualmente, 10 de 11 pacientes viven con una mediana de seguimiento de 3,1 años (rango: 1-5,7). La curva de Kaplan-Meier obtiene una supervivencia global del 90,9% y libre de eventos de 81,8% (fig. 3).
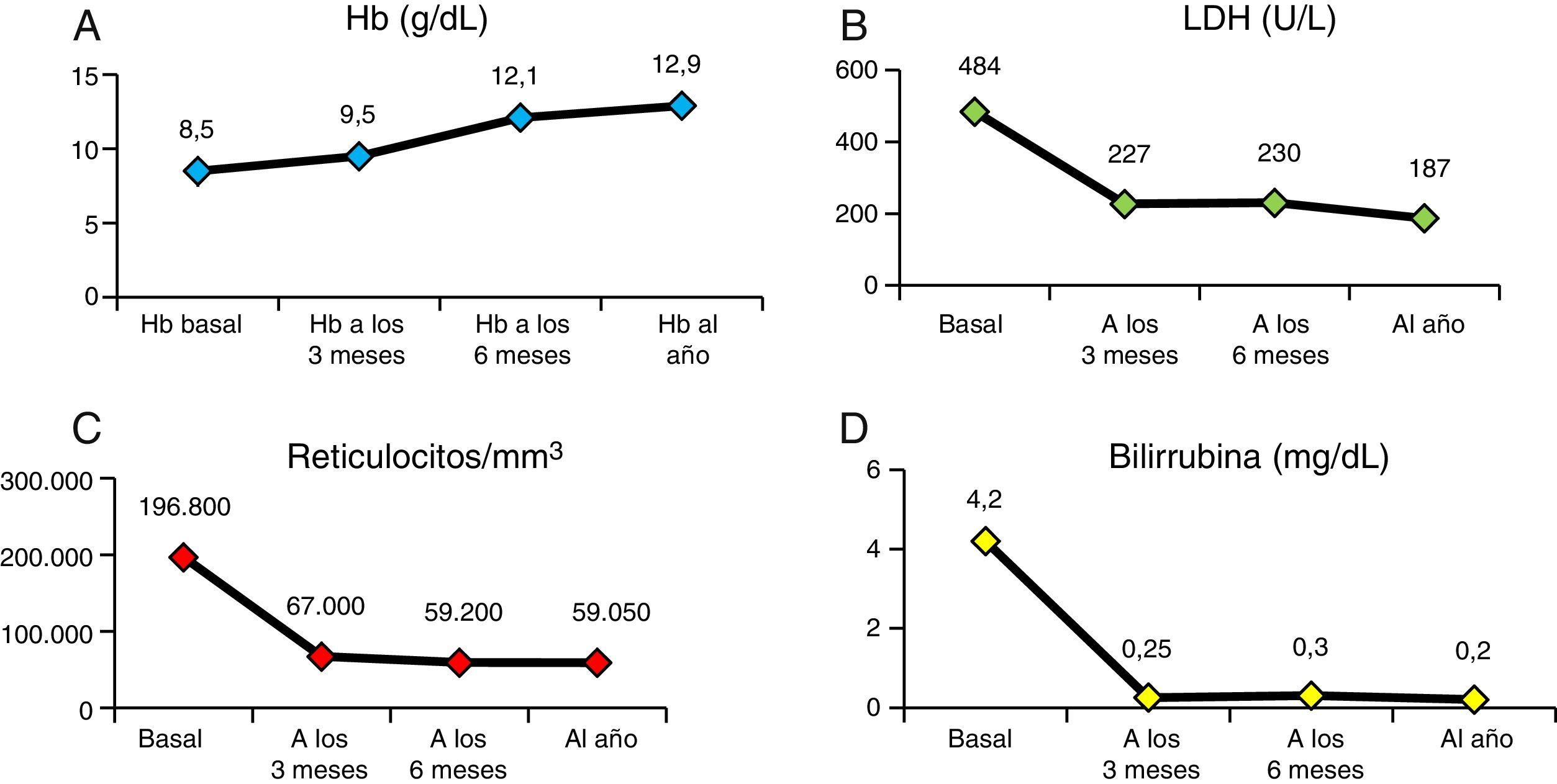
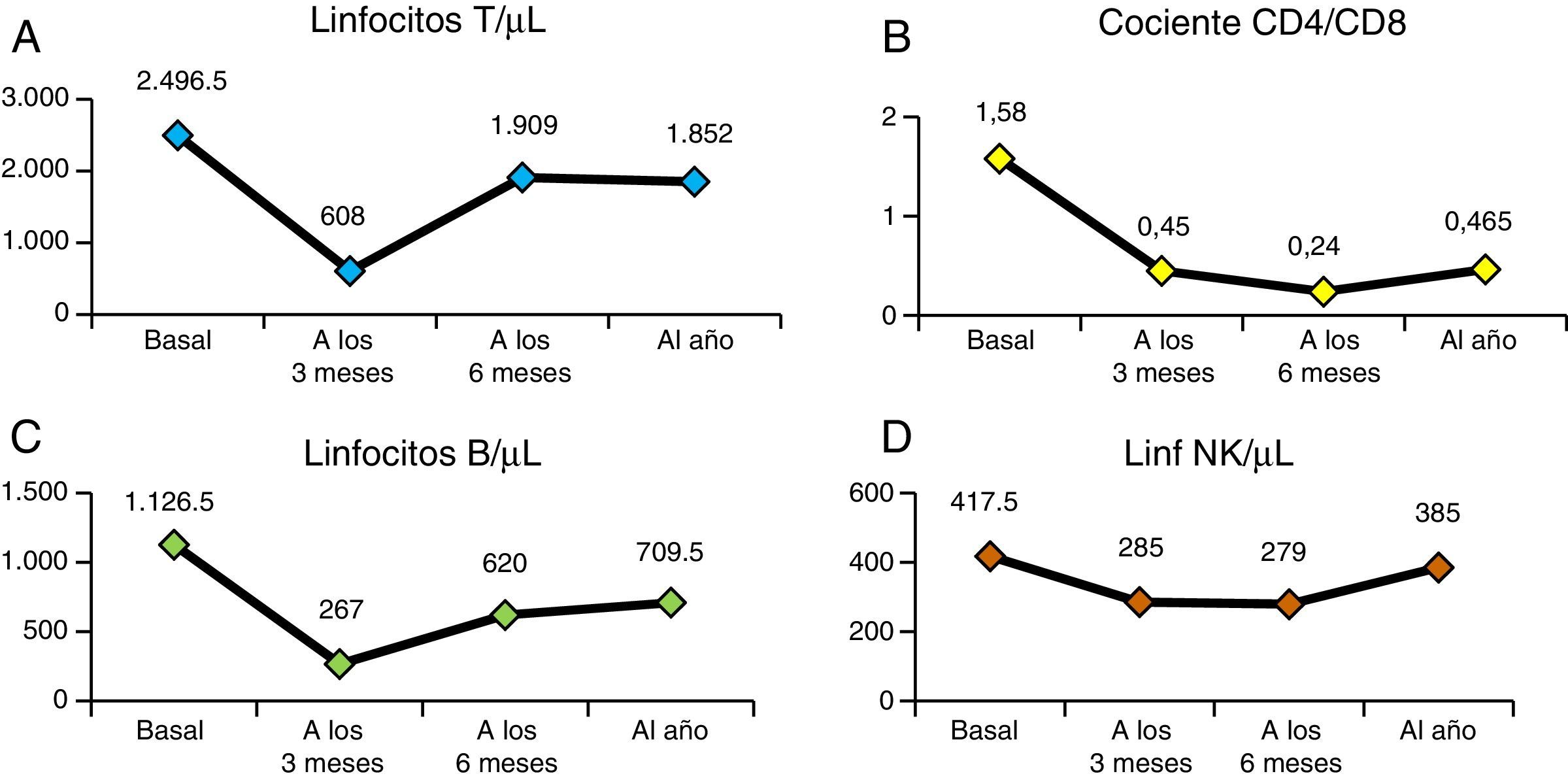
En la figura 4 vemos la evolución de los parámetros de hemólisis (hemoglobina, reticulocitos, LDH y bilirrubina), existiendo diferencia significativa (p<0,05) entre las medianas del valor basal de los pacientes y al año del TPH. En la figura 5 aparece representada la evolución de la reconstitución inmune (linfocitosT, B y NK, cociente de linfocitos T CD4/CD8) con las medianas del valor basal, a los 3, 6 meses y al año del TPH.
En nuestro estudio, pese al número limitado de pacientes (n=11) se obtienen resultados acordes con los estudios multicéntricos8,9,15 en países con más experiencia en el tratamiento de niños con ECF. El procedimiento del TPH en estos pacientes implica no solo un conocimiento de la técnica del trasplante, sino un profundo conocimiento del tratamiento general de las hemoglobinopatías. El acceso de nuestro país a cohortes de niños con ECF es relativamente reciente, por lo que esta serie es la más amplia publicada.
Un paciente fallece a consecuencia de EICHa, una de las principales causas de muerte también en otros estudios (tabla 3). La incidencia de EICHa (> grado ii) es del 36%, mayor que en otras series pese a que la profilaxis es similar mediante ciclosporina y metotrexato, aunque estos datos podrían modificarse con un mayor tamaño muestral. Una posible causa es que la fuente de progenitores hematopoyéticos en todos nuestros casos es la médula ósea, a diferencia de los otros estudios, donde hasta un 14% proceden de cordón umbilical, con menor riesgo de EICHa14,16,17. Sin embargo, ningún paciente en nuestra serie desarrolla EICHc.
Experiencia internacional. Comparación del estudio actual con series previamente reportadas
| Estudio actual | Vermylen 1998 | Walters 2000 | Bernaudin 2007 | Panepinto 2007 | |
|---|---|---|---|---|---|
| N.° | 11 | 50 | 50 | 87 | 67 |
| Supervivencia global | 90.9% | 93% | 94% | 93.1% | 98% |
| Supervivencia libre de eventos | 81.8% | 82% | 84% | 86.1% | 85% |
| Fallo injerto | 9% | 10% | 10% | 7% | 13% |
| EICHa (≥ grado ii) | 36% | 20% | 15% | 20% | 10% |
| EICHc | 0% | 20% | 12% | 13.5 | 22% |
| N.° muertes/causa | 1/EICH | 2: EICH+CMV+Aspergillus Muerte súbita | 3/EICH | 6: 4: EICH Hemorragia SNC Sepsis | 3: EICH+Hemorragia SNC Fallo multiorgánico Causa desconocida |
La vasculopatía cerebral es causa de importante morbilidad neurológica relacionada con el trasplante en los pacientes con ECF. Walter et al.18 describe una incidencia de complicaciones neurológicas post-TPH del 50% en pacientes con antecedentes de ictus y del 23% en el resto de pacientes; sin embargo, en la serie francesa14 el antecedente de enfermedad cerebrovascular no se demostró un factor de riesgo significativo. En nuestro caso, los 2 pacientes con ACV en la RM previa al TPH no desarrollan complicaciones neurológicas agudas. La HTA aparece en 7 de 11 pacientes y causa importante morbilidad puesto que, pese a las medidas preventivas, se asocia probablemente en 3 pacientes con HSA. La incidencia de crisis tanto en la serie francesa como americana es de un 24 y un 28%, respectivamente, siendo la complicación neurológica post-TPH más frecuente1; estas se producen con mayor frecuencia en los primeros días post-TPH y en relación con HTA y con medicación como corticoides14,18. En nuestro estudio, pese a profilaxis con fenitoína, 3/11 pacientes presentan crisis, todos ellos en relación con HTA. El paciente 1 presenta en el día +27 de nuevo crisis, en esta ocasión relacionada con toxicidad por ciclosporina, presentando como secuela déficit cognitivo leve. No existe en nuestra serie mortalidad relacionada con complicaciones neurológicas ni secuelas neurológicas graves.
El fallo de injerto y la reconstitución autóloga permanecen como otros de los principales problemas en el TPH en los pacientes con ECF, con una incidencia que oscila entre el 7 y el 13% (tabla 3) y en nuestro estudio un 9%. Entre las causas se encuentran la aloinmunización previa por múltiples transfusiones, la presencia de una médula ósea hiperregenerativa, la incompatibilidad de grupo, la celularidad (CD34+) infundida y las infecciones víricas. La serie francesa demostró que el aumento de inmunosupresión mediante introducción de timoglobulina de ratón en el régimen de acondicionamiento disminuye la probabilidad de rechazo desde un 22,6 a un 3%14. Todos nuestros pacientes reciben alemtuzumab como fármaco antilinfocitario. En el paciente 9 se produce una reconstitución autóloga con reaparición de la clínica de ECF en el día +180 y como factores de riesgo destacan una celularidad infundida en el límite bajo, una incompatibilidad mayor de grupo y una reactivación del CMV (tablas 1 y 2). La retirada de la inmunosupresión e infusión de linfocitos T de donante no resulta de utilidad en nuestro paciente para mejorar el quimerismo que, aunque en el día +30 es completo en médula ósea total, ya presenta el 51% de linfocitos T del receptor. En el paciente 11, al igual que en el 9, se objetiva quimerismo mixto en la población de linfocitos T en el día +30, por lo que se decide cambio de estrategia con aumento de la inmunosupresión añadiendo micofenolato a la ciclosporina, con lo que mantiene un quimerismo estable al año de seguimiento y sin manifestaciones clínicas de la ECF (tabla 2). El quimerismo mixto puede ser una opción en las patologías hematológicas no malignas y aunque su principal riesgo es el desarrollo de reconstitución autóloga en el primer año, si permanece estable permite evitar el desarrollo de clínica relacionada con la enfermedad19 y la mejoría de los parámetros de hemólisis20.
A raíz de los resultados obtenidos en los estudios multicéntricos, nuevas estrategias surgen con la intención de disminuir la mortalidad relacionada con el trasplante, el fallo de injerto y la toxicidad ocasionada. Así un estudio realizado en Bélgica obtiene una mejoría significativa de la supervivencia libre de eventos (ningún caso de fallo de injerto o reconstitución autóloga) en el grupo que recibe hidroxiurea en los meses previos al TPH, frente al grupo que recibe el acondicionamiento estándar con busulfán, ciclofosfamida y ATG, sin hidroxiurea (97,4% vs. 66,7%, p=0,006)21.
Recientemente, sobre la base de estudios en adultos con acondicionamientos de toxicidad reducida y un estudio en niños con talasemia mayor22, se ha empleado en 15 niños con ECF un acondicionamiento con treosulfán, fludarabina y tiotepa. El treosulfán tiene una estructura análoga al busulfán, con gran actividad mieloablativa pero con menor toxicidad extramedular y una farmacocinética más lineal. En dicho estudio, se obtienen excelentes resultados con una SG de 100% y SLE de 93% y con menor toxicidad relacionada con el TPH alentando al cambio de acondicionamiento en estos pacientes23.
En nuestro estudio, todos los pacientes de la serie tenían indicación para TPH según la guía de práctica clínica de ECF de la SEHOP1; sin embargo, los mejores resultados se obtienen en los pacientes trasplantados en fases precoces de la enfermedad, como se demostró en la serie belga donde la supervivencia global y libre de eventos de los pacientes asintomáticos era superior a la de aquellos trasplantados con formas graves (100% vs. 88% y 93% vs. 76%, respectivamente)13. Es por ello que en la actualidad se plantea el debate de realizar el TPH de hermano HLA idéntico en pacientes asintomáticos o ampliando las indicaciones24.
ConclusiónEl TPH es la única opción curativa en pacientes con ECF y nuestros resultados son acordes con las publicaciones internacionales (SG 90,9%, SLE 81,82%). Sin embargo, persiste el riesgo de muerte (9%), siendo el EICH su causa principal e importantes comorbilidades como el fallo de injerto (9%) y las complicaciones neurológicas (36%). A la vista de estos resultados y de las publicaciones referidas, se ha decidido el cambio del acondicionamiento (treosulfán, fludarabina, tiotepa), la supresión de la eritropoyesis en los 2 meses previos al TPH, la monitorización previa mediante MAPA de la PA, y se contempla el cambio en la profilaxis del EICH. El estudio del quimerismo en linfocitos T resulta de utilidad en la detección precoz del quimerismo mixto y, en nuestra experiencia, con mejores resultados tras el aumento de inmunosupresión como medida para evitar el fallo de injerto.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
