







Sickle cell disease (SCD), despite the improvement in the medical management, is still associated with severe morbidity and decreased survival. Allogenic hematopoietic stem cell transplantation (Allo-HSCT) currently provides the only curative therapy. A report is presented on our experience in children with SCD, who underwent Allo-HSCT in a single centre.
Material and methodA single centre descriptive study was conducted on patients with SCD who underwent a bone marrow transplant from an HLA-identical sibling donor between January 2010 and December 2014. Epidemiological, clinical and analytical parameters were collected with a follow-up to December 2015. Data are presented as frequencies, percentages, and medians (range).
ResultsAllo-HCST was performed in 11 patients (8 males) with a median age of 7 years (2–13), all of them with comorbidity prior to the HCST. A stable graft was achieved in 10 out of 11 patients, 9 of them with complete donor chimerism, and one patient with stable mixed chimerism after 1 year of allo-HSCT. One patient has secondary graft failure with re-appearance of symptoms associated with SCD on day 180. Complications of Allo-HSCT are: arterial hypertension 7/11, acute renal failure 3/11, CMV reactivation 9/11, neurological complications 4/11 (subarachnoid haemorrhage, seizure), and acute graft versus host disease (aGVHD) of the skin 6/11, one of whom developed grade IV intestinal aGVHD, causing his death (day 51). None of the patients developed chronic GVHD. The overall survival and event-free survival was 90.9% and 81.9%, respectively, with a median follow-up of 3.1 (1–5.7) years.
ConclusionsAllo-HSCT, the only curative therapy, remains associated with morbidity. There was a transplant related mortality in our study, consistent with multicentre studies (1/11), and with aGVHD being the main cause. Other problems still include graft failure (1/11), and neurological complications (4/11), although the permanent sequelae are mild.
La enfermedad de células falciformes (ECF), pese a la mejora en el manejo médico, persiste asociada a morbilidad y a menor supervivencia. El alotrasplante de progenitores hematopoyéticos (alo-TPH) es actualmente la única opción curativa. Describir la evolución clínico-analítica de los pacientes trasplantados en nuestro centro.
Material y métodoEstudio unicéntrico descriptivo, incluye a pacientes con ECF en los que se realizó alo-TPH de médula ósea de hermano HLA-idéntico desde enero del 2010 hasta diciembre del 2014. Se recogen datos epidemiológicos, clínicos y analíticos con tiempo de seguimiento hasta diciembre del 2015. Los datos se presentan como frecuencias, porcentajes y medianas (rango).
ResultadosSe recluta a 11 pacientes (8 varones), mediana de edad: 7 años (2–13), todos ellos con comorbilidad previa al TPH. Se consigue injerto estable en 10/11 pacientes, quimerismo completo en 9/11 y quimerismo mixto estable tras un año del TPH en 1/11. Un paciente presenta fallo secundario de injerto con reaparición de clínica el día +180. Complicaciones post-TPH: complicaciones neurológicas 4/11 pacientes (hemorragia subaracnoidea, crisis), HTA 7/11, fallo renal agudo 3/11, reactivación CMV 9/11, EICHa cutáneo 6/11, uno de ellos desarrolla EICH intestinal grado IV causando su fallecimiento (día +51). Ningún paciente desarrolla EICH crónico. Supervivencia global y libre de eventos a los 3,1 años de seguimiento: 90,9 y 81,9%, respectivamente.
ConclusionesEl alo-TPH, única opción curativa, no está exento de morbimortalidad, encontramos un riesgo de muerte similar a otras series (1/11), siendo su primera causa el EICH agudo. Otros problemas son fallo de injerto (1/11) y complicaciones neurológicas (4/11), aunque las secuelas permanentes son leves.
Sickle cell disease (SCD) is currently the abnormality detected most frequently in newborn screening tests in several countries, including Spain. In the Autonomous Community of Madrid, where screening has been performed since May 2003, this is corroborated with an incidence of 1 in every 5000 live births.1 Sickle cell disease is a significant source of multisystemic morbidity and carries an increased risk of early death.2 In the past three decades, the introduction of several measures, such as penicillin prophylaxis, vaccination against Streptococcus pneumoniae, the use of hydroxyurea, have been implemented leading to a decrease in morbidity and mortality in patients with SCD.3–5 There is evidence that hydroxyurea considerably reduces the incidence of vaso-occlusive events and acute chest syndrome6,7; however, this treatment does not reverse organ damage or reduce the risk of stroke. The use of transcranial Doppler ultrasonography (TCD) in the followup of these patients allows the identification of those that are at high risk of experiencing an acute stroke, while of brain magnetic resonance imaging (MRI) allows the diagnosis of silent strokes.8 Transfusion therapy has proven to be useful in the primary prevention of stroke, but it has the disadvantage of secondary iron overload and poor long-term adherence.9
At present, haematopoietic stem cell transplantation (HSCT) is the only available curative treatment. The first successful HSCT was performed in 1984 in the Untied States in a patient with SCD and acute myeloid leukaemia.7 Subsequent multicentre studies have reported good outcomes, with an overall survival of 93–94% and an event-free survival of 82–86%, while the most frequent complications are graft failure and graft-versus-host disease (GVHD).10–14
The aim of our study was to describe the clinical and analytical outcomes of patients with SCD that underwent allogeneic HSCT from an HLA-identical sibling donor in our hospital.
Material and methodsWe conducted a retrospective descriptive single-centre study. We collected data for paediatric patients with homozygous (HbSS) or double heterozygous (HbSβ0) SCD that underwent bone marrow transplantation from an HLA-identical sibling donor (Hb AA or Hb AS phenotype) between January 2010 and December 2014, with followup until December 2015. We studied demographic variables (age, sex) and variables concerning the patients’ history of complications associated with the disease: acute chest syndrome (ACT), vaso-occlusive crises (VOCs), dactylitis, priapism, stroke, splenic sequestration and need for splenectomy. As regards treatment prior to HSCT, we documented whether patients received hydroxyurea (HU), transfusion therapy or iron chelation therapy. Other variables under study associated with HSCT were donor-recipient ABO compatibility, stem cell infusion, day of platelet and neutrophil engraftment and chimerism outcomes in bone marrow at day +30 post transplant and in peripheral blood later in time. We collected the following laboratory parameters measured at baseline and at 3, 6, 9 and 12 months post transplantation: haemoglobin, reticulocytes, lactase dehydrogenase (LDH), bilirubin, ferritin, glomerular filtration rate based on cystatin C, lymphocyte differential count (B and T lymphocytes, NK cells), and immunoglobulins (Igs). Last of all, we gathered data for post-transplant complications: acute GVHD, neurologic complications (seizures, subarachnoid haemorrhage [SAH], neurotoxicity), acute renal failure, hypertension (HTN), cytomegalovirus (CMV) reactivation and cause of death.
Conditioning regimenThe conditioning regimen was myeloablative, and consisted of intravenous busulfan (dose by weight of 3.2–4.8mg/kg/day, administered as a single dose every 24h on days −9 to −6), intravenous alemtuzumab (0.1mg/kg/day on days −8 to −6) and intravenous cyclophosphamide (50mg/kg/day, on days −5 to −2, combined with mesna). Prophylaxis treatment against GVHD was administered from day −1 and consisted of intravenous ciclosporin for three to six months and two doses of methotrexate (at days +4 and +7). Polymerase chain reaction was used to assess for the presence of CMV and EBV and antibody testing to assess for Aspergillus, and infection prophylaxis treatment given consisting of micafungin, levofloxacin and acyclovir, as well as cotrimoxazol after engraftment. Additional care measures included intravenous immunoglobulin therapy, ursodiol, phenytoin, antihypertensive drugs to maintain blood pressure at approximately the 95th percentile, supplementation with magnesium as needed and transfusion support with irradiated blood products. The procedure was carried out in individual rooms with positive pressure ventilation systems with HEPA filters.
All patients underwent monthly transfusions or exchange transfusions to achieve a level of Hb S of less than 30% before HSTC, and during transplantation, platelet counts were maintained at more than 50000units/μL and Hb concentrations at 11–13g/dL. Infusion of haematopoietic stem cells was always done from fresh bore marrow. In donor 1, due to the difference in weight with the recipient, bone marrow was extracted in the months preceding transplantation and cryopreserved until infusion (day +1), 24h after the fresh bone marrow infusion.
Statistical analysisWe used the SPSS® software to analyse the data. We have expressed the data as frequencies, percentages, mean and medians (ranges). We compared paired quantitative data by means of Wilcoxon's test, and considered results with p-values of less than 0.05 statistically significant.
We studied overall survival and even-free survival by the Kaplan and Meier method. We defined event as death, graft loss (chimerism>95% of recipient cells) or recurrence of SCD.
DefinitionsWe defined neutrophil and platelet engraftment as the first of three consecutive days with total counts of more than 500neutrophils/μL or more than 50000platelets/μL. We defined full donor chimerism as more than 95% of hematopoietic cells of donor origin.
We defined primary graft failure as failure to achieve by day +28 post transplantation a neutrophil count greater than 500μL−1, platelet count greater than 50000μL−1 and a haemoglobin concentration greater than 8g/dL, and secondary graft failure as the lost of at least two cell lineages that had been functional after initial engraftment. We defined graft rejection or autologous reconstitution as recurrence of SCD symptoms.
ResultsPatient characteristics (Table 1)From January 2010 to December 2014, 11 patients (8 male and 3 female) underwent allogeneic HSCT from the bone marrow of an HLA-identical sibling. All of them had a homozygous haemoglobin S phenotype (Hb SS). The median age was 7 years (2–13). Pathological manifestations before HSCT included recurrent VOCs (n=9), recurrent ACS (n=6), stroke (n=2), abnormal flow velocity in TCD (n=4), splenectomy (n=2) and testicular atrophy (n=1). The baseline treatment prior to HSCT consisted of hydroxyurea in four patients, transfusion therapy in one, and hydroxyurea and periodic transfusions in three. Two patients developed iron overload and required iron chelation therapy (Table 1).
Characteristics of patients and donors.
| Patient | Sex | Age of BMT | Hydroxyurea TX | BMT indication | Comorbidities | Donor | ||
|---|---|---|---|---|---|---|---|---|
| Hb type | Recipient/donor ABO group | Recipient/donor CMV IgG | ||||||
| Pt 1 | M | 13 y, 10 m | Yes | Abnormal TCD, VOC, ACS | Hb AA | A Rh+/O Rh+ | +/+ | |
| Pt 2 | F | 7 y, 10 m | No | Stroke, abnormal TCD | Moya-Moya syndrome | Hb AA | AB Rh+/AB Rh- | +/+ |
| Pt 3 | F | 10 y, 5 m | Yes | Abnormal TCD, VOC | Hb AA | A Rh+/B Rh+ | +/+ | |
| Pt 4 | M | 5 y, 6 m | Yes | VOC, ACS | Testes: atrophy (right), microcalcifications (left) | Hb AA | A Rh+/A Rh + | +/+ |
| Pt 5 | M | 9 y, 8 m | Yes | Stroke, abnormal TCD, VOC | Hb AS | B Rh+/O Rh+ | +/+ | |
| Pt 6 | F | 4 y, 9 m | No | ACS, VOC | Hb AS | B Rh+/B Rh+ | +/+ | |
| Pt 7 | M | 9 y, 2 m | Yes | ACS, VOC | Hb AS | O Rh+/O Rh+ | +/+ | |
| Pt 8 | M | 2 y, 7 m | No | ACS, VOC | Hb AA | A Rh+/A Rh+ | −/+ | |
| Pt 9 | M | 9 y, 10 m | Yes | ACS | Splenectomy. Left ventricular noncompaction | Hb AS | O Rh+/B Rh– | +/− |
| Pt 10 | M | 2 y, 1 m | No | VOC | Hb AS | A Rh+/O Rh+ | +/+ | |
| Pt 11 | M | 6 y, 1 m | Yes | VOC | Splenectomy | Hb AS | O Rh+/O Rh+ | −/+ |
ACS, acute chest syndrome; BMT, bone marrow transplant; y, years; m, months; F, female; M, male; TCD, transcranial Doppler; TX, treatment; VOC, vaso-occlusive crisis.
All bone marrow donors were HLA-identical siblings; 55% had an Hb AS phenotype. There was minor blood group incompatibility in four patients and major incompatibility in two.
Graft and post-haematopoietic stem cell transplantation complications (Table 2)A stable graft was achieved in ten out of eleven patients, nine of who developed full donor chimerism (Table 2).
Haematopoietic stem cell transplantation (HSCT), outcomes and complications post transplantation.
| Pt | Cell dose: CD34+ (×106kg−1) | Day NEU engraftment | Day Plat engraftment | Chimerism (% recipient origin) | CMV (day and treatment) | |
|---|---|---|---|---|---|---|
| Day +30 (BM) | Last chimerism (time post HSCT) | |||||
| Pt 1 | Day 0 (fresh BM): 3.8 Day +1 (cryopreserved BM): 6.1 | +16 | +31 | 1.9% | 4.4% (5 y 7 m) | Yes, +9, foscarnet |
| Pt 2 | 6 | +23 | +27 | 0% | 4.8% (4 y 8 m) | Yes, +13, foscarnet |
| Pt 3 | 3.4 | +23 | +27 | 0% | 3.6% (4 y 7 m) | Yes, +11, foscarnet |
| Pt 4 | 8.5 | +27 | +27 | 0% | 3.6% (4 y 1 m) | No |
| Pt 5 | 5.48 | +16 | +38 | 0% | 0% (3 y 1 m) | Yes, +13, foscarnet |
| Pt 6 | 4.63 | +18 | +42 | 0% | 0% (3 y 9 m) | Yes, +7, foscarnet |
| Pt 7 | 3.72 | +20 | +19 | 0% | Yes, +39, ganciclovir | |
| Pt 8 | 6.52 | +25 | +39 | 0% | 0% (1 y 9 m) | Yes, +47, ganciclovir |
| Pt 9 | 2.85 | +18 | +28 | 1.3% 51% T cells | 92% 88% T cells (1 y 7 m) | Yes, +8, foscarnet |
| Pt 10 | 5.3 | +24 | +94 | 2.4% | 0% (1 and 2 m) | Yes, +9, foscarnet |
| Pt 11 | 3.5 | +22 | +24 | 2.5% 59% T cells | 6% 12% T cells (1 y 0 m) | No |
| Pt | Acute neurologic complications (day of onset) | HTN (day of onset) | AKF | aGVHD-localization and grade | Other complications post-HSCT | Event (death/graft loss/SCD clinical recurrence/alive) | |
|---|---|---|---|---|---|---|---|
| Seizures | SAH | ||||||
| Pt 1 | Yes, +1 +27 | Yes, +1 | Yes (+1) | No | Yes Cutaneous II | Day +27: brain MRI: ciclosporin toxicity Mild cognitive impairment Mucositis grade 3 | Alive |
| Pt 2 | No | No | Yes (day 0) | No | No | Alive | |
| Pt 3 | Yes, +29 | No | Yes (+17) | No | Yes Cutaneous I | Alive | |
| Pt 4 | No | No | No | No | No | Mucositis grade 4 | Alive |
| Pt 5 | No | No | Yes (+13) | Yes | Yes Cutaneous II | Possible Aspergillus infection | Alive |
| Pt 6 | No | No | No | No | No | Catheter-related infection and left gluteus myositis due to MRSA | Alive |
| Pt 7 | Yes, +1 | Yes, +1 | Yes (+1) | Yes | Yes Cutaneous IV Gastrointestinal IV Hepatic III | BK virus-associated haemorrhagic cystitis Cholestasis and development of biliary sludge and cholelithiasis Fever without microbial isolation | Died on day +51 (aGVHD grade IV and multiple organ failure) |
| Pt 8 | No | No | No | No | Yes Cutaneous I | Mucositis grade 4 | Alive |
| Pt 9 | No | Yes, +2 | Yes (day −1) | No | No | Mucositis grade 1 BK virus-associated haemorrhagic cystitis | Clinical recurrence (VOC) on day +180 |
| Pt 10 | No | No | Yes (+23) | Yes | Yes Cutaneous III | Mucositis grade 3 Fever and neutropaenia | Alive |
| Pt 11 | No | No | No | No | No | Mixed chimerism: intensified immunosuppression for 1 year | Alive |
BM, bone marrow; CMV, cytomegalovirus; aGVHD, acute graft-versus-host disease; AKF, acute kidney failure; BM, bone marrow; HTN, hypertension; NEU, neutrophil; y, years; m, months; PB, peripheral blood; Plat, platelet; SCD, sickle cell disease; SAH, subarachnoid haemorrhage; VOC, vaso-occlusive crisis.
The median neutrophil engraftment was 22 days (16–27) and the median platelet engraftment was 28 (19–94).
Patient 9 (Table 2) had full donor chimerism in the overall bone marrow at day +30 (1.3% cells of recipient origin), but chimerism analysis of cell subpopulations showed that 51% of T lymphocytes were of recipient origin. Immunosuppression was discontinued and the patient was given three infusions of donor T lymphocytes at days +71 (1×105kg−1), +105 (1.95×105kg−1) and +170 (0.85×106kg−1) with no improvement in chimerism, and the symptoms of SCD recurred on day +180 (admission for VOC). Patient 11 (Table 2) had full donor chimerism in the overall bone marrow at day +30 (2.5% cells of recipient origin) but partial chimerism in the T lymphocyte population (59% of recipient origin). Immunosuppression was intensified, adding mycophenolate to ciclosporin and maintaining the treatment for one year. The mixed chimerism values remained stable: 4.2% at three months, 6% at six months, 6% at one year, with a gradual decrease of recipient T lymphocytes (12% at one year).
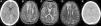
The most frequent complication after HSCT was reactivation of CMV, which occurred in nine out of eleven patients and was treated with foscarnet when it developed prior to engraftment and valganciclovir if the graft was stable. Hypertension developed in seven out of eleven children in association with stem cell infusion (patients 1 and 7), secondary to administration of drugs such as corticosteroids for the treatment of acute GVHD (patient 3), foscarnet for reactivation of CMV (patient 5) or ciclosporin (patient 9), or due to multiple factors in the context of impaired renal function (patient 10). All patients achieved adequate control of blood pressure, save for patient 10, who still needs oral hypertensive treatment one year later, although blood pressure has normalised and treatment is being tapered off. Fig. 1 shows the evolution of glomerular filtration assessed by measurement of cystatin C in patients that developed acute kidney failure.
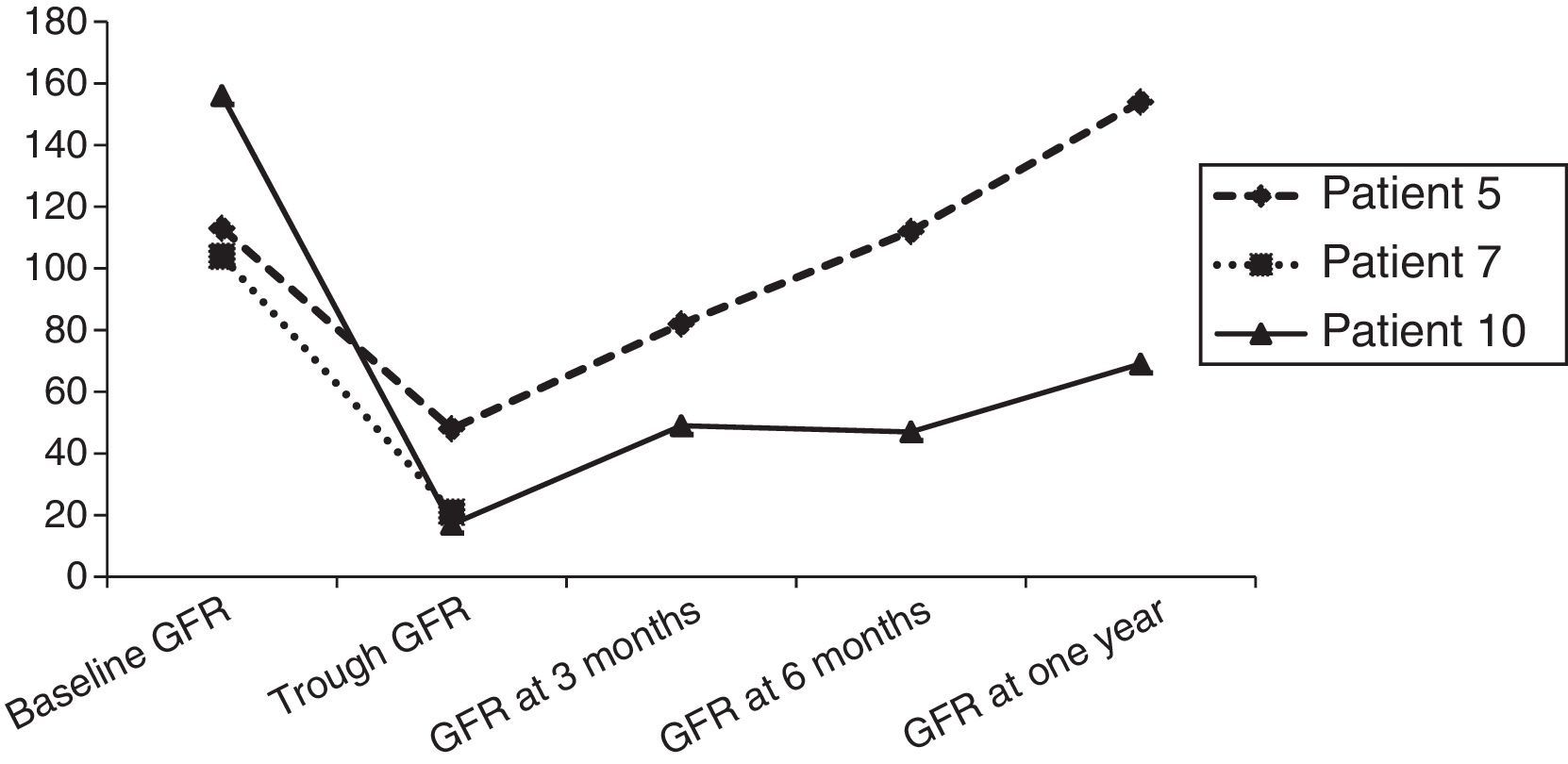
When it came to neurologic complications, three patients developed SAH in the context of hypertension, two of who had associated epileptic seizures (Table 2 and Fig. 2). Patient 1 had a second episode of seizures on day +27, and computed tomography (CT) did not detect new sites of bleeding, although it showed bilateral parieto-occipital and frontal hypodense lesions. The investigation was expanded with brain MRI, which ruled out vascular malformations and found lesions in both cerebral hemispheres compatible with ciclosporin toxicity and development of oedema post transplantation. The patient had a favourable neurologic outcome (cognitive and motor) after discontinuation of ciclosporin, although a mild cognitive impairment remained. A follow-up MRI scan at six months revealed the persistence of the previously observed lesions, with no new lesions (Fig. 2). The neurologic evaluation of patients that had SAH at one year post transplantation was normal, and while patient 1 experienced academic difficulties (mild cognitive impairment), he did not require grade retention and has college-level studies.
Patient 1 (A–D). Simple axial CT scan with sulcus hyperdensity suggestive of subarachnoid haemorrhage in the right lateral sulcus, superolateral sulci with right-side predominance, and the left peripeduncular cistern (A). T2 TSE axial MRI at day +27 with evidence of bilateral frontal cortical and subcortical lesions and bilateral frontal and left parietal deep white matter lesions compatible with ciclosporin toxicity (B and C). T2 TSE axial MRI at six months post HSCT with evidence of residual lesions (D). Patient 9 (E): axial head CT scan with hyperdensity in left frontotemporal sulci as evidence of subarachnoid haemorrhage on day +2.
Six patients developed acute GVHD, five with isolated skin involvement that responded well to corticosteroid treatment. Patient 7 had acute GVHD refractory to corticosteroids with grade IV skin and intestinal involvement and grade III liver involvement that did not respond to intensification of immunosuppression (mycophenolate, infliximab) or photopheresis, leading to his death on day +51. None of the patients developed chronic GVHD or hepatic sinusoidal obstruction syndrome.
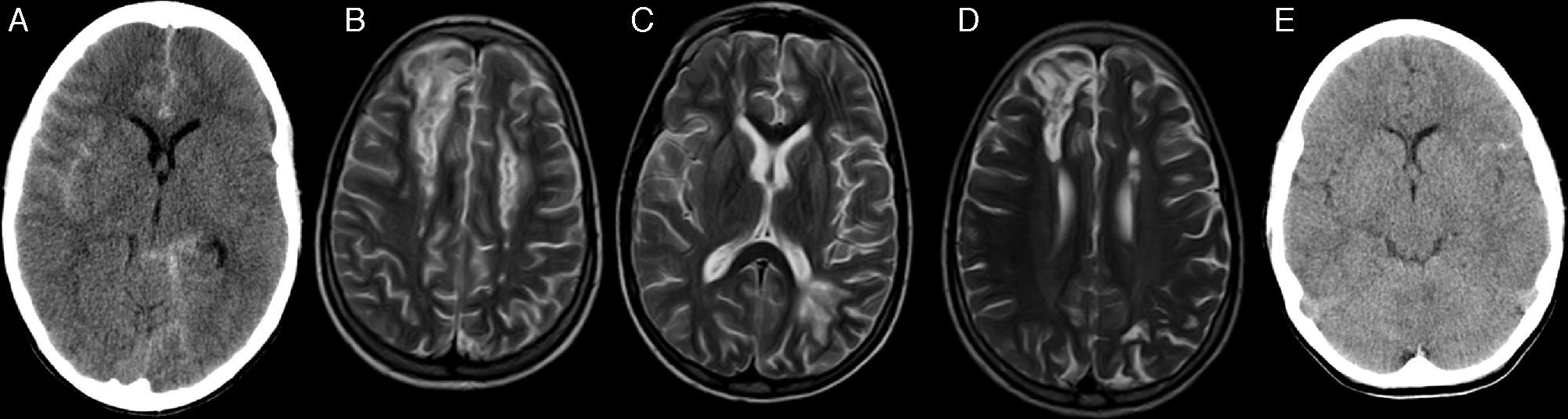
At the time of this writing, 10 out of the 11 patients are alive after a mean duration of followup of 3.1 years (range, 1–5.7). The Kaplan–Meier curve shows an overall survival of 90.9% and an event-free survival of 81.8% (Fig. 3).
Fig. 4 presents the evolution of haemolysis parameters (haemoglobin, reticulocytes, LDH and bilirubin), with significant differences (P<.05) between the median patient levels at baseline and at one year after HSCT. Fig. 5 presents graphs of the evolution of immune reconstitution (T and B lymphocytes, NK cells, T CD4/CD8 ratio) with the median counts at baseline and at three months, six months and one year post transplantation.
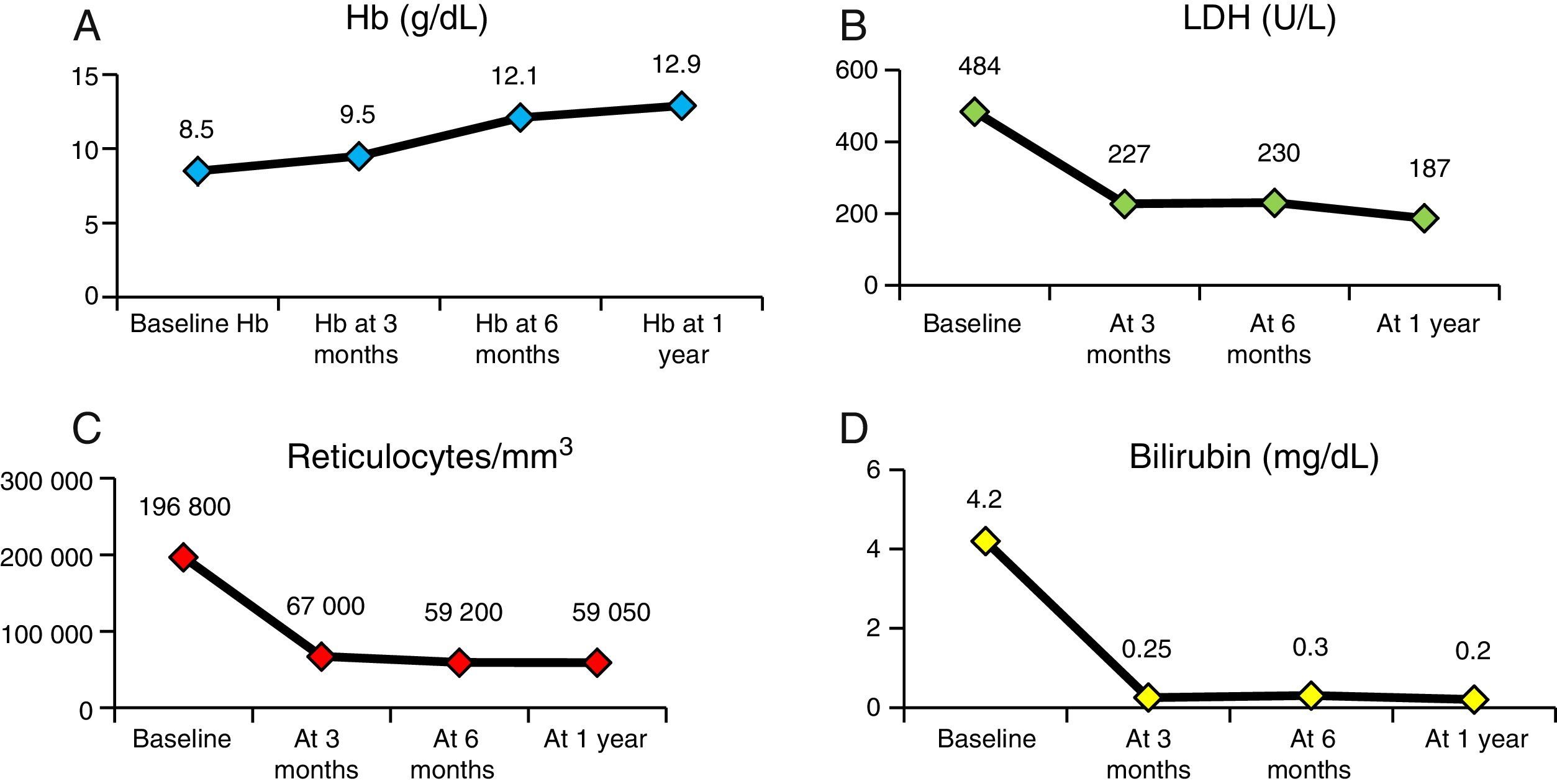
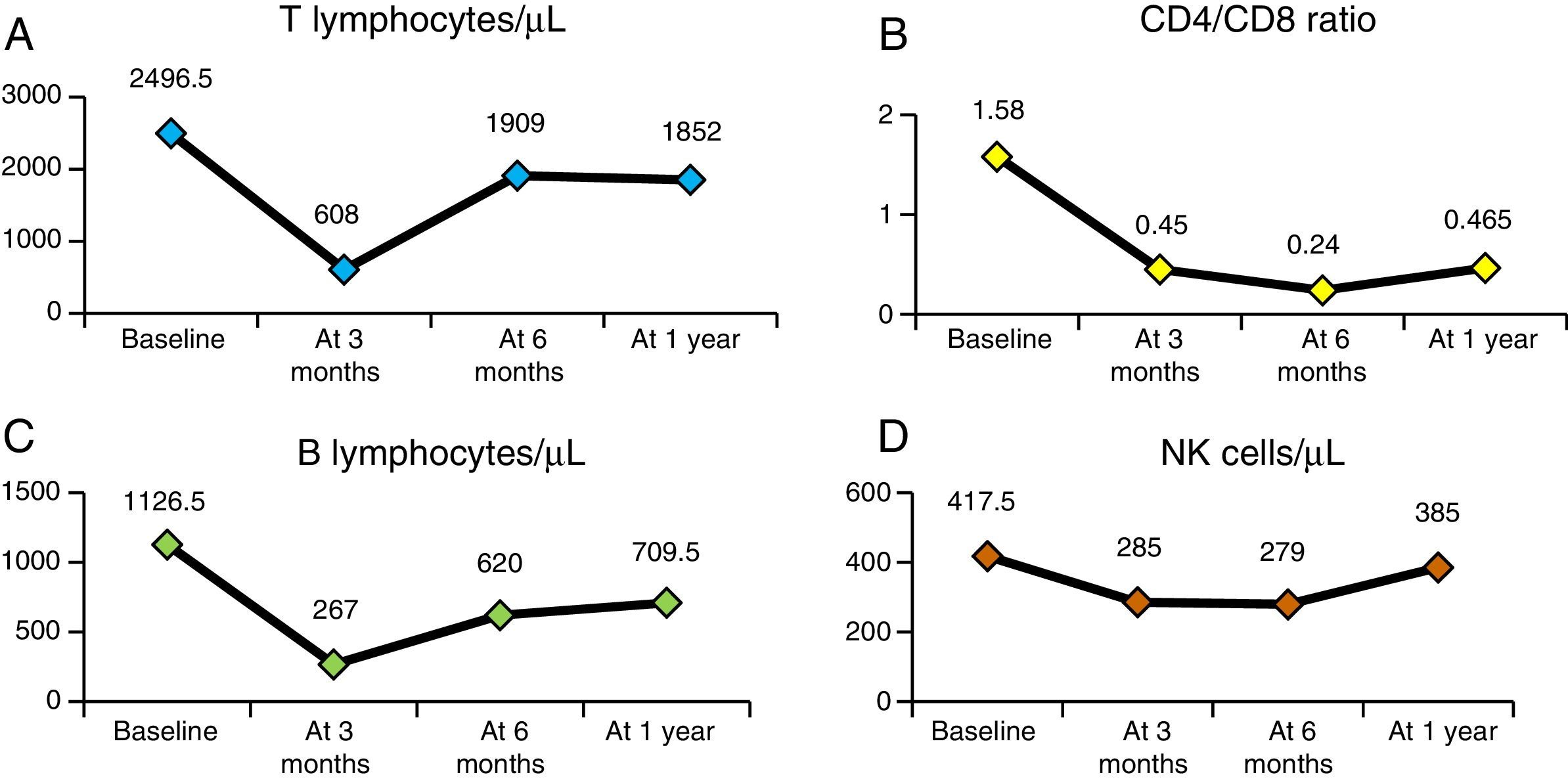
Despite the small sample size (n=11), the results of our study were consistent with those of multicentre studies8,9,15 conducted in countries with more experience in the treatment of children with SCD. Performance of HSCT in these patients requires not only knowledge of transplantation technique, but also a thorough understanding of the overall treatment of haemoglobinopathies. In Spain, access to cohorts of children with SCD is a relatively new phenomenon, so this is the largest series studied to date.
One patient died of acute GVHD, which is also one of the main causes of death in other series (Table 3). The incidence of acute GVHD (grade>II) was 36%, higher than the incidence reported in other series despite similar prophylaxis regimens with ciclosporin and methotrexate, although this result may have been different with a larger sample size. A possible cause is that the source of the stem cells was bone marrow in all cases, whereas in other studies the source is umbilical cord blood, which carries a lower risk of acute GVHD, in up to 14% of cases.14,16,17 At any rate, none of the patients in our series developed chronic GVHD.
International experience. Comparison of the current study with previously published series.
| Current study | Vermylen 1998 | Walters 2000 | Bernaudin 2007 | Panepinto 2007 | |
|---|---|---|---|---|---|
| N | 11 | 50 | 50 | 87 | 67 |
| Overall survival | 90.9% | 93% | 94% | 93.1% | 98% |
| Event-free survival | 81.8% | 82% | 84% | 86.1% | 85% |
| Graft failure | 9% | 10% | 10% | 7% | 13% |
| aGVHD (≥ grade ii) | 36% | 20% | 15% | 20% | 10% |
| cGVHD | 0% | 20% | 12% | 13.5 | 22% |
| No. deaths/cause | 1/GVHD | 2: GVHD+CMV+Aspergillus Sudden death | 3/GVHD | 6: 4: GVHD CNS haemorrhage Sepsis | 3: GVHD+CNS haemorrhage Multiple organ failure Unknown cause |
aGVHD, acute graft-versus-host disease; cGVHD, chronic graft-versus-host disease.
Cerebrovascular problems are a significant cause of neurologic morbidity associated with transplantation in patients with SCD. Walter et al.18 reported an incidence of neurologic complications post HSCT of 50% in patients with a prior history of cerebral infarction and of 23% in the rest of the patients; however, a French series13 did not find a significant increase in risk in patients with a past history of cerebrovascular disease. In our series, the two patients with evidence of stroke in the MRI scan performed before HSCT did not develop acute neurologic complications. Hypertension developed in seven out of the eleven patients and caused significant morbidity, as despite the measures taken for its management, it was probably related to SAH in three patients. The incidence of seizures in the French and North American series was 24% and 28%, respectively, and seizures are the most frequent complication post HSCT1; they usually develop in the first few days after transplantation in association with hypertension and with corticosteroid therapy.13,18 In our study, and despite prophylaxis with phenytoin, three of the eleven patients developed seizures, all of them in association with hypertension. Patient 1 had a new episode of seizures on day +27, this time due to ciclosporin toxicity, and leading to mild cognitive impairment. In our series, patients did not die from neurologic complications nor develop severe neurologic sequelae.
Graft failure and autologous reconstitution also continue to be among the major problems that follow HSCT in patients with SCD, with an incidence that ranges between 7% and 13% (Table 3) and of 9% in our study. Among its causes are previous alloimmunisation due to multiple transfusions, bone marrow hyper-regeneration, blood group incompatibility, cell dose (CD34+) and viral infection. The French series demonstrated that intensification of immunosuppression with addition of rabbit anti-thymocyte globulin to the conditioning regimen reduces the probability of graft rejection from 22.6% to 3%.13 All our patients received alemtuzumab to induce lymphocyte depletion. Patient 9 experienced autologous reconstitution with recurrence of SCD symptoms on day +180, with the main risk factors for it being a cell dose near the lower limit, major blood group incompatibility and reactivation of CMV (Tables 1 and 2). Discontinuation of immunosuppression and infusion of donor T lymphocytes did not succeed in improving chimerism in our patient, who had full donor chimerism in the overall bone marrow at day +30, but with 51% of T lymphocytes already of recipient origin. Similarly, patient 11 had mixed chimerism in the T lymphocyte population at day +30, which led to a change in management, intensifying immunosuppression with the addition of mycophenolate to ciclosporin; this had achieved a stable chimerism at one year of followup in the absence of clinical manifestations of SCD (Table 2). Mixed chimerism can be an acceptable option in nonmalignant blood disorders, and although the main risk it carries is autologous reconstitution in the first year, if it remains stable it can prevent the development of clinical manifestations of the disease19 and improve haemolysis parameters.20
The findings of multicentre studies have led to the development of new management strategies with the purpose of reducing transplant-related mortality, graft failure and treatment toxicity. Thus, a study conducted in Belgium found a significant improvement in event-free survival (no cases of graft failure or autologous reconstitution) in the group treated with hydroxyurea in the months preceding HSCT, compared to the group treated with the standard conditioning regimen with busulfan, cyclophosphamide and anti-thymocyte globulin and no hydroxyurea (97.4% vs. 66.7%; P=.006).21
Recently, based on the results of studies in adults treated with reduced-toxicity conditioning regimens and one study in children with thalassaemia major,22 a study in 15 children with SCD used a conditioning regimen consisting of treosulfan, thiotepa and fludarabin. Treosulfan has a structure analogous to that of busulfan and exhibits a potent myeloablative activity combined with minimal extramedullary toxicity and a more linear pharmacokinetic profile. This study obtained excellent results, with an overall survival of 100%, an event-free survival of 93% and a decreased treatment-related toxicity for HSCT, encouraging a change in the conditioning of these patients.23
In our study, HSCT was indicated in all patients based on the clinical practice guidelines of SCD of the SEHOP1; however, patients that receive the transplant in the early stages of the disease have the best outcomes, as was observed in the Belgian series, in which overall survival and event-free survival of asymptomatic patients were higher than those of patients with severe forms of disease (100% vs. 88% and 93% vs. 76%, respectively).12 This is why there is an ongoing debate on whether HSCT from an HLA-identical sibling donor should be performed in asymptomatic patients or its indications should be expanded.24
ConclusionHaematopoietic stem cell transplantation is the only curative treatment available to patients with SCD, and our results are consistent with those of the international literature (overall survival, 90.9%, event-free survival, 81.82%). However, it still carries a risk of death (9%), with the most frequent reason being GVHD, and of significant comorbidities, such as graft failure (9%) and neurologic complications (36%). In light of these results and the reviewed literature, we decided to make changes to the conditioning regimen (treosulfan, fludarabin, thiotepa), the suppression of erythropoiesis in the two months prior to HSCT and pre-transplantation ambulatory blood pressure monitoring, and are contemplating a change in GVHD prophylaxis. T lymphocyte chimerism analysis helps the early detection of mixed chimerism and, in our experience, leads to better outcomes through the subsequent intensification of immunosuppression to prevent graft failure.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: García Morin M, Cela E, Garrido C, Bardón Cancho E, Aguado del Hoyo A, Pascual C, et al. Trasplante de médula ósea en pacientes con anemia falciforme. Experiencia en un centro. An Pediatr (Barc). 2017;86:142–150.
