



El síndrome de Mayer-Rokitansky-Küster-Hauser (SMRKH) se caracteriza por la aplasia congénita del útero y los dos tercios superiores de la vagina en mujeres con un desarrollo adecuado de los caracteres sexuales secundarios y un cariotipo normal 46,XX1. La incidencia se estima entre 1:4.000-5.000 mujeres nacidas vivas2. El síndrome de Jarcho-Levin (SJL) o disostosis espondilocostal se define por la asociación de malformaciones costales y vertebrales, condicionando acortamiento troncular y talla baja3. Afecta a 1:4.000 nacidos vivos4. A continuación presentamos el caso de una paciente con diagnóstico neonatal de SJL que consulta por amenorrea primaria, poniendo de manifiesto una agenesia útero-vaginal.
Niña de 15 años con amenorrea primaria. Sin antecedentes prenatales de interés, diagnosticada al nacimiento de SJL por presentar tronco corto y malformaciones toracolumbares. Asocia también comunicación interventricular perimembranosa, comunicación interauricular tipo ostium secundum y hernia inguinal derecha con inclusión de ovario.
En el momento de su valoración presenta un desarrollo puberal completo (estadio de Tanner V), genitales externos femeninos normales. No se objetivan signos de hiperandrogenismo. Para estudiar la amenorrea primaria se solicita ecografía pélvica, edad ósea (EO) y estudio hormonal. En la ecografía no se evidencian útero ni ovarios. La EO es acorde a su edad cronológica. La analítica muestra estradiol (92ng/ml) y gonadotropinas (LH 13,1mUI/ml y FSH 4,6mUI/ml) en rango normal. Se realiza una resonancia magnética abdomino-pélvica (fig. 1) en la que no se identifica útero ni tercio superior de vagina. Los ovarios estan en posición normal y en el derecho se demuestra un quiste. Los riñones tienen morfología y posición normal.
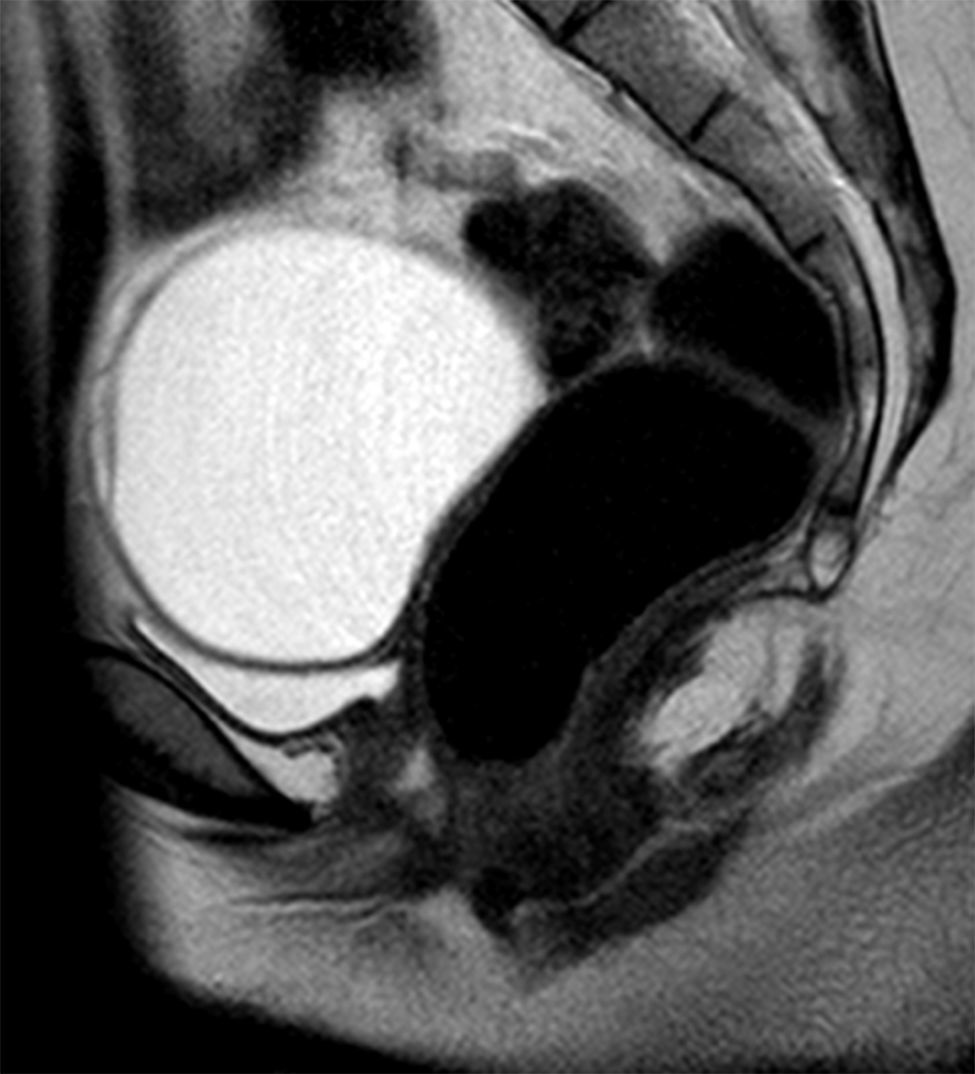
Resonancia magnética pélvica. Imagen sagital potenciada en T2 en la que se identifica un quiste de ovario improntando el techo de la vejiga y se puede identificar que la pared posterior de la vejiga y la anterior del recto están en contacto por la ausencia de útero y tercio superior de vagina.
Ante tales hallazgos la paciente es diagnosticada de SMRKH, el cual puede ser de 2 tipos: el tipo I (OMIM 277000) o aplasia útero-vaginal aislada y el tipo II (OMIM 601076), que como corresponde con el caso que presentamos se asocia a otras anomalías congénitas, principalmente renales, cardíacas, auditivas y esqueléticas. A nivel esquelético se describen defectos vertebrales (aislados, escoliosis, Klippel-Feil), costales, malformaciones de paladar o de extremidades1.
Dado el origen mesodérmico común y la estrecha relación embriológica de los órganos afectados se propone que el SMRKH puede originarse en fases muy tempranas del desarrollo embrionario. La existencia de casos familiares con diferente grado de afectación lleva a buscar posibles genes candidatos como el que codifica para la hormona anti-mülleriana (AMH) y su receptor, Wt1, PAX2, los genes homeobox HOX y WNT, sin que se demuestre una relación causal. Se describen casos de SMRKH tipo II asociado a hiperandrogenismo en pacientes con mutaciones en WNT41,5. A su vez, el SJL se asocia a defectos del gen DLL3, que forma parte de la vía de señalización Notch, implicada en el desarrollo embriológico3. La presencia de diferentes locus en los genes que pueden relacionarse con ambas entidades, resulta sugestivo de que en este caso probablemente exista un origen multifactorial, poligénico o que la causa sea una alteración común a varias vías de señalización implicadas.
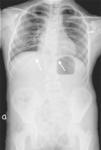
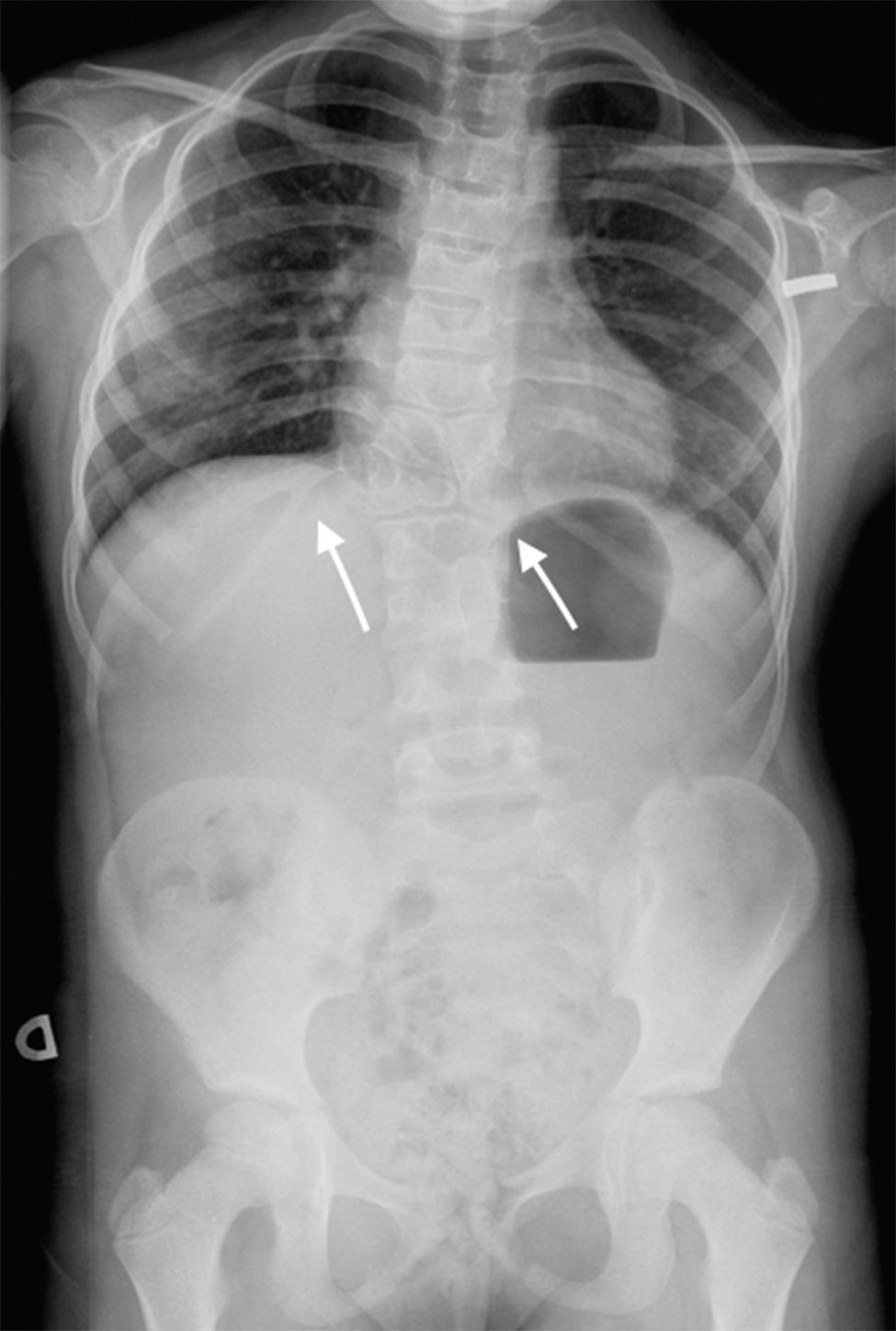
Nuestra paciente inicialmente se diagnostica de SJL dada la presencia de tórax en escudo, T12 en mariposa, fusión vertebral L1-L2-L3, y anomalías costales (fusión de la undécima y duodécima costilla derecha, anomalías morfológicas de segunda y tercera costilla derecha y ausencia de la primera costilla derecha) (fig. 2). Posteriormente, dada la ausencia de útero y porción superior de la vagina se une el diagnóstico de SMRKH. Actualmente la paciente presenta escoliosis no progresiva con curva dorsal derecha de 45°, patrón respiratorio restrictivo moderado y no tiene lesiones cardíacas residuales; mantiene seguimiento multidisciplinar.
Para las mujeres con SMRKH, las posibilidades de ser madres se reducen a la adopción, la gestación subrogada o el trasplante uterino. Fruto de esta última técnica nace el primer bebé vivo en 20146, por otro lado, la bioingeniería suscita un interés creciente en el tratamiento de la infertilidad femenina, con el desarrollo potencial de tejidos y órganos sustitutivos, que entre otras ventajas evitarían la necesidad de inmunosupresión.
Con la comunicación de este caso queremos presentar la primera asociación entre el SMRKH y el SJL.
Si bien se describen anomalías vertebrales y/o costales asociadas al SMRKH, en la bibliografía revisada no encontramos ningún caso con características de SJL o disostosis espóndilo-costal. Así mismo, en algún paciente con SJL se asocian alteraciones urogenitales, pero ninguna paciente de las descritas en la bibliografía cumple criterios de SMRKH.
En este caso se detectan de forma precoz malformaciones óseas y cardíacas, sin embargo se llega a un diagnóstico tardío del SMRKH, lo cual puede evitarse realizando un despistaje completo de otras anomalías.
