



El síndrome Schinzel-Giedion (SSG) (#MIM 269150) es una enfermedad genética infrecuente, caracterizada por dismorfia cráneo-facial específica, anomalías congénitas múltiples y discapacidad intelectual grave. La mayoría de los pacientes fallece en los primeros años de vida. Se debe a mutaciones en el gen SETBP1, habiéndose descrito a la fecha un reducido número de pacientes con confirmación molecular.
Presentamos a un paciente de 4 años con SSG asociado a la mutación c.2608G>T (p.Gly870Cys) en el gen SETBP1, no descrita previamente. Se revisan las características clínicas de esta enfermedad y su diagnóstico diferencial.
Los rasgos dismórficos son muy característicos en el SSG. Su reconocimiento clínico es fundamental para alcanzar un diagnóstico precoz, planificar un correcto seguimiento y ofrecer asesoramiento genético familiar adecuado. A la fecha, este es el decimoséptimo paciente publicado con mutación en el gen SETBP1, primero en España, contribuyendo a ampliar el conocimiento clínico y molecular de esta entidad.
Schinzel-Giedion syndrome (SGS) (#MIM 269150) is a rare genetic disorder characterized by very marked craniofacial dysmorphism, multiple congenital anomalies and severe intellectual disability. Most affected patients die in early childhood. SETBP1 was identified as the causative gene, but a limited number of patients with molecular confirmation have been reported to date.
The case is reported of a 4 and a half year-old male patient, affected by SGS. SETBP1 sequencing analysis revealed the presence of a non-previously described mutation: c.2608G>T (p.Gly870Cys). The clinical features and differential diagnosis of this rare condition are reviewed.
Dysmorphic features are strongly suggestive of SGS. Its clinical recognition is essential to enable an early diagnosis, a proper follow-up, and to provide the family with genetic counseling. To date, this is the seventeenth SGS patient published with SETBP1 mutation, and the first in Spain, helping to widen clinical and molecular knowledge of the disease.
El síndrome Schinzel-Giedion (SSG; #MIM 269150) es una enfermedad genética infrecuente, caracterizada por discapacidad intelectual grave, dismorfia craneofacial y anomalías congénitas múltiples, incluyendo alteraciones esqueléticas, genitourinarias y cardiacas, entre otras1, así como un aparente aumento del riesgo de neoplasias de estirpe embrionaria y neuroepitelial2-6. Fue descrito en 1978 por Schinzel y Giedion7 y, desde entonces, se han comunicado 52 casos. En 2010, Hoischen et al.8 describieron mutaciones en heterocigosis, de novo, en el gen SETBP1 (ubiquitous SET-binding protein, *611060, 18q21.1), identificándolo como responsable de esta entidad. A la fecha, tan solo 16 pacientes con mutación han sido publicados8-11. Presentamos a un nuevo paciente con SSG, primero en España, con una nueva mutación en SETBP1.
Caso clínicoVarón de 4 años y medio, segundo hijo de padres sanos, no consanguíneos. Gestación natural con detección de hidronefrosis, micropene, agenesia de cuerpo calloso y pabellones auriculares displásicos en ecografía de semana 27. Cesárea programada en semana 38. Apgar 8/9. Peso: 3.480 g (p79). Longitud: 50,5cm (p72). PC: 35cm (p75). Ingreso al nacimiento en hospital de origen. Su hemograma, bioquímica, gasometría, amonio y cribado metabólico fueron normales. Los potenciales auditivos demostraron hipoacusia bilateral mixta. El ecocardiograma descartó cardiopatía. La ecografía abdominal confirmó hidronefrosis bilateral grado iii, y la cistoureterografía detectó reflujo vesicoureteral (RVU) bilateral grado iii-iii. La RM cerebral mostró hipoplasia de cuerpo calloso y dilatación ventricular. La serie ósea informó de displasia epifisaria múltiple, sin calcificaciones patelares. El cariotipo fue normal. Planteada la sospecha diagnóstica, de síndrome de Robinow, la secuenciación del gen ROR2 no identificó mutaciones.
Desde los 3 meses, el paciente es atendido en nuestro hospital por crisis convulsivas iniciadas al mes de vida, con diagnóstico de encefalopatía grave, con práctica ausencia de adquisiciones en todas las áreas y epilepsia sintomática.
Remitido a Genética Médica, la exploración física a los 3 años mostraba un peso de 10,5kg (p2, –2,27 DE), talla 86cm (p < 1, –3,51 DE) y PC: 48cm (p2, –2,06 DE). Braqui-turricefalia. Pelo ralo y fino. Hipertricosis. Aspecto tosco. Frente amplia. Sutura metópica prominente. Cejas pobladas. Hipertelorismo. Proptosis. Surco palpebral inferior. Hipoplasia mediofacial. Puente nasal aplanado. Nariz corta. Narinas antevertidas. Labios finos. Protrusión lingual. Retrognatia. Pabellones auriculares de implantación baja, con lóbulos gruesos. Aumento de distancia intermamilar. Micropene. Encefalopatía grave. Limitación a la extensión de rodillas, dedos de manos y codos. Pliegue palmar único derecho. Braquidactilia generalizada y clinodactilia de 5.° dedo de las manos.
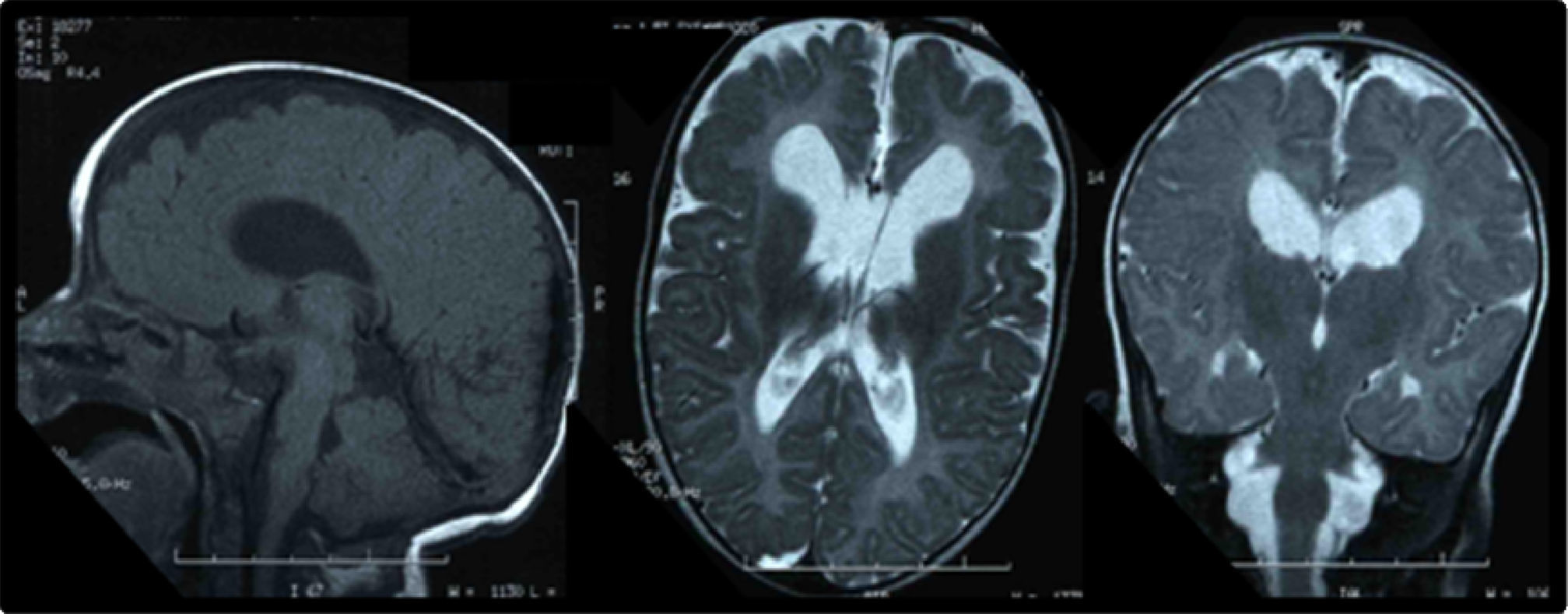
El estudio metabólico resultó normal (aminoácidos, ácidos orgánicos, saicar, sulfitest, biotinidasa, lactato/piruvato, acilcarnitinas, esteroles, sialotransferrinas, ácidos grasos poliinsaturados, de cadena muy larga, pristánico y fitánico). El electroencefalograma de larga duración detectó irritación cerebral paroxística polifocal de predominio izquierdo. Una nueva RM cerebral evidenció áreas de paquigiria fronto-basal y polimicrogiria bitemporal (fig. 1). En la evaluación oftalmológica, los medios oculares fueron transparentes y el fondo de ojo normal, hallando obstrucción del conducto lacrimonasal. Los potenciales visuales demostraron alteración grave, mayor en el ojo derecho. El FISH (fluorescence in situ hybridization) 17p13 (síndrome de Miller-Dieker) y el MLPA (multiplex ligation-dependent probe amplification) de subtelómeros resultaron normales. Con la sospecha clínica de SSG, se realizó secuenciación del gen SETBP1 (Hôpital Trousseau, París), identificando la mutación c.2608G>T (p.Gly870Cys) en heterocigosis en el exón 4, no descrita con anterioridad, de novo.
El paciente ha presentado retraso pondoestatural desde los primeros meses de vida, con dificultad para la alimentación, vómitos frecuentes y estreñimiento pertinaz. Ante reflujo gastroesofágico con neumonías espirativas, precisó técnica antirreflujo de Nissen y sonda de gastrostomía.
En su seguimiento nefrourológico, la gammagrafía renal con DMSA (ácido dimercaptosuccínico marcado con Tc99) renal mostró función del 50% en ambos riñones. Evolutivamente, ha presentado infecciones urinarias de repetición, precisando STING.
En seguimiento por Neuropediatría, en tratamiento con valproato y lamotrigina, con buen control de crisis. Ha recibido tratamiento de las contracturas articulares con toxina botulínica y ortesis. Mantiene programa de estimulación y fisioterapia, con mejoría en la interacción social, tono postural, sostén cefálico y de tronco, desplazamiento reptando y, recientemente, deambulación con andador.
DiscusiónEl SSG se caracteriza por dismorfia craneofacial específica, destacando estrechamiento bitemporal, hipoplasia mediofacial, órbitas prominentes, protrusión lingual e hipertricosis7. El fenotipo se modifica en el tiempo12,13. Como se aprecia en nuestro paciente, los rasgos toscos persisten, pero la hipertricosis disminuye y la hipoplasia mediofacial se hace menos evidente (fig. 2). Esta evolución fenotípica puede dificultar el diagnóstico a edades más avanzadas, pudiendo ser útil la valoración de fotografías con menor edad.
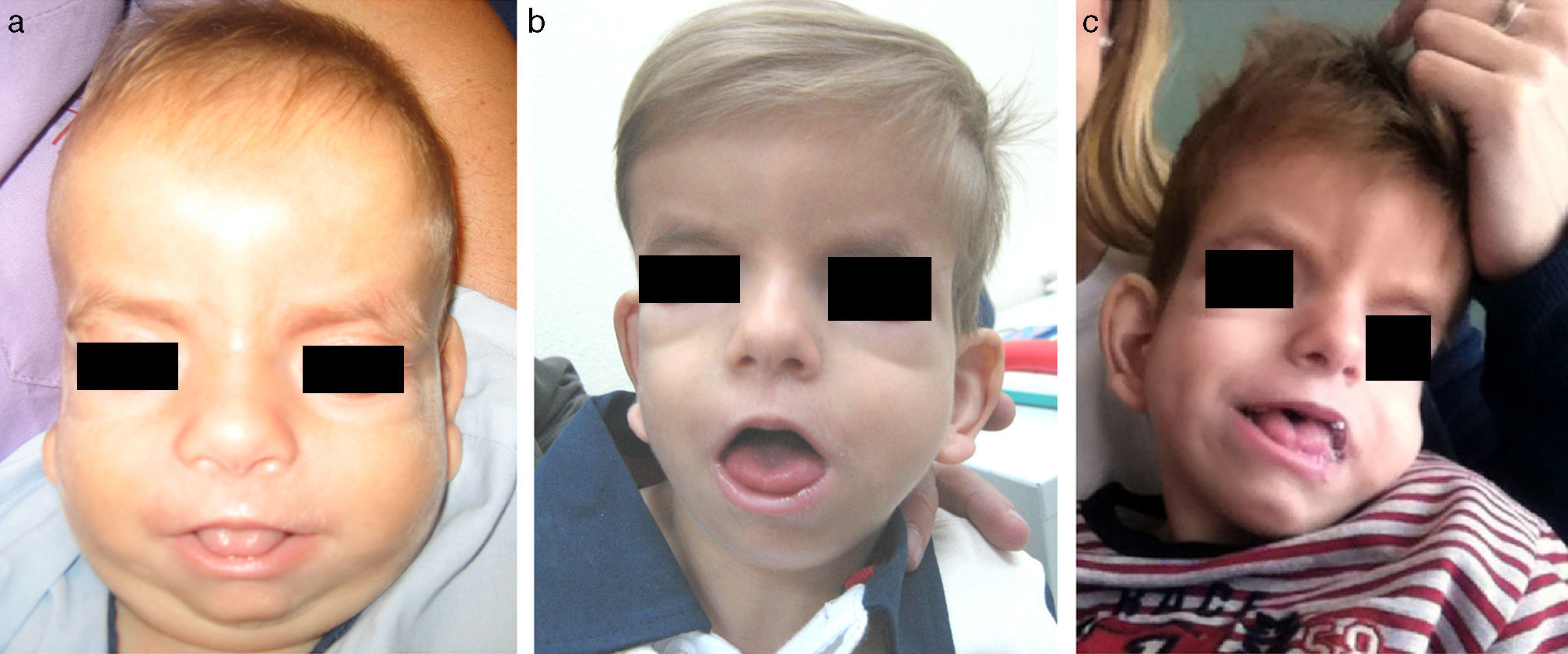
a) Paciente a los 6 meses. Cráneo con morfología en «8», frente amplia, hipertelorismo, proptosis, pliegue palpebral inferior, hipoplasia mediofacial, nariz corta e hipertricosis. b) Paciente a los 3 años. Evolución fenotípica con disminución de la hipertricosis y la retracción mediofacial. c) Paciente a los 4 años y medio. Rasgos menos toscos. Sutura metópica prominente. Disminución del relleno periorbitario y la proptosis. Persistencia de glosoptosis.
El aspecto tosco de los pacientes recuerda al hipotiroidismo o enfermedades metabólicas de depósito14, llevando a plantear en nuestro paciente inicialmente la posibilidad de síndrome de Zellweger. Por otro lado, la presencia de turricefalia, frente alta, nariz corta con narinas antevertidas y encefalopatía malformativa, recordaban en los primeros meses el síndrome de Miller-Dieker. En el hospital de origen, se planteó posibilidad de síndrome de Robinow, si bien el fenotipo del paciente no es compatible con este diagnóstico. En la literatura, el diagnóstico diferencial incluye el síndrome de Okamoto15, con discapacidad intelectual, rasgos dismórficos, hipertricosis e hidronefrosis congénita, entre otros. En nuestra opinión, una vez conocida esta entidad, es clínicamente distinguible de todas las previamente expuestas. Por tanto, su reconocimiento, pese a su escasa frecuencia, es de crucial importancia para alcanzar un diagnóstico precoz, disminuyendo la incertidumbre familiar y el gasto diagnóstico.
Las anomalías esqueléticas están presentes en más del 90% de los pacientes e incluyen sincondrosis occipital, huesos wormianos, costillas anchas, clavículas largas, braquimelia, sinostosis radio-cubital, engrosamiento de diáfisis de huesos largos, tibias arqueadas e hipoplasia de falanges distales, entre otros9. A nivel locomotor, en nuestro paciente destaca la limitación a la extensión articular. La serie ósea a los 3 meses informó de ausencia de anomalías craneales significativas, aumento del diámetro antero-posterior de cuerpos vertebrales dorsales, leve ensanchamiento de región lateral de arcos costales y de región medial de ambas clavículas, ramas púbicas cortas y leve hipoplasia de falanges distales (fig. 3). Las características radiológicas constituyen, por tanto, una importante ayuda diagnóstica16.
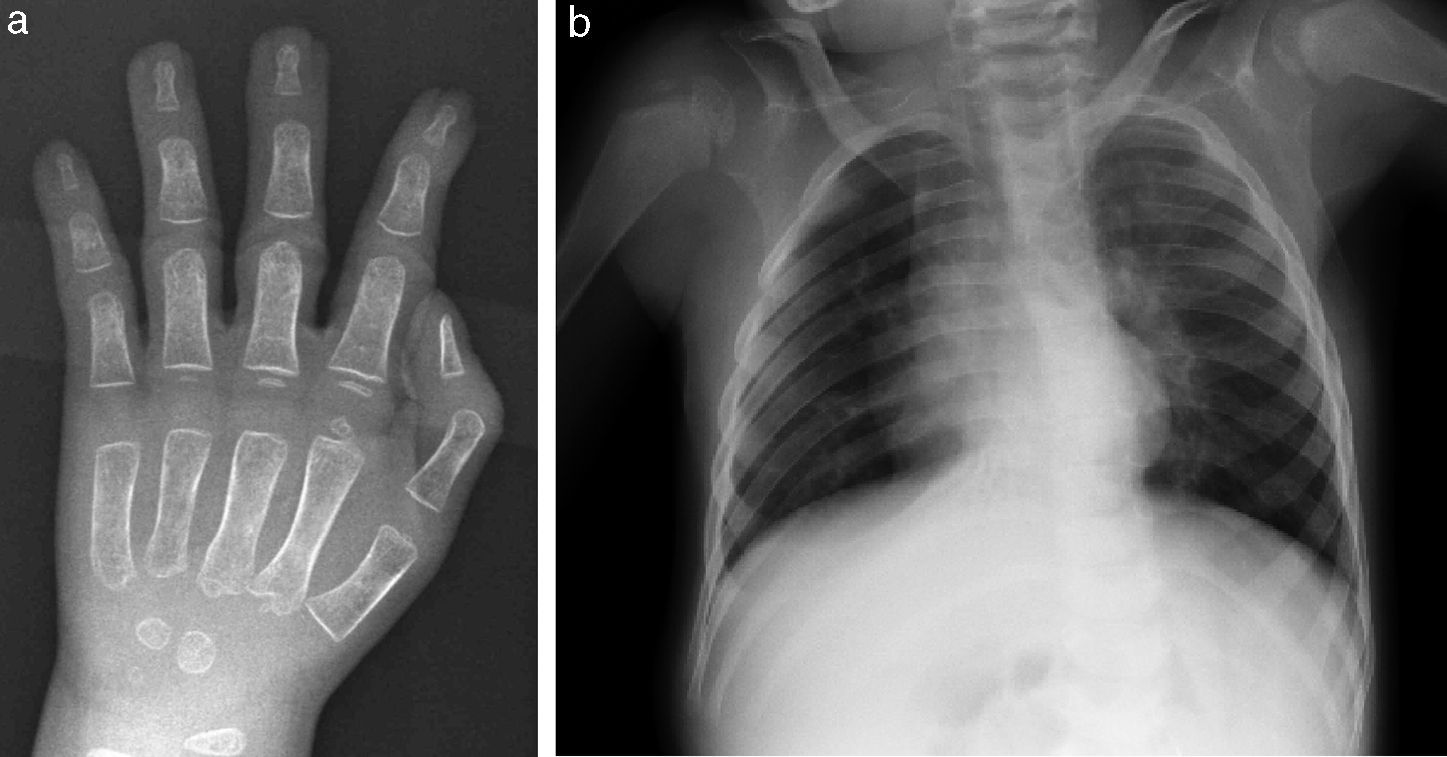
Las anomalías genitourinarias están presentes en el 90% de los pacientes. Al igual que en nuestro caso, la mayoría de los pacientes presentan megacaliosis e hidronefrosis congénita. Esta malformación se ha descrito asociada a trisomía 13, trisomía 18, síndrome de. Turner, triploidías, síndrome de Johansson-Blizzard y síndrome de Ochoa. Por tanto, su detección supone igualmente un importante signo guía para el diagnóstico13.
Los pacientes presentan discapacidad intelectual grave y espasticidad. Hasta el 73% asocia epilepsia17. En nuestro caso, el comienzo se produjo al mes de vida, con respuesta favorable al tratamiento antiepiléptico.
Entre los hallazgos de neuroimagen, se incluyen atrofia cerebral, aumento del tamaño ventricular, hipoplasia-agenesia de cuerpo calloso, quistes aracnoideos y trastorno de migración neuronal, entre otros12,18, como la paquigiria y la polimicrogiria presentes en nuestro paciente. Se ha descrito mielinopatía vacuolizante evolutiva, similar a la observada en otras enfermedades (hiperglucinemia no cetósica, enfermedad de Canavan, enfermedad del jarabe de arce, fenilcetonuria y deficiencia de metionina adenosiltransferasa i/iii), indicando la posibilidad de neurodegeneración asociada14,17,19.
Nuestro paciente presenta déficit visual y auditivo, hallazgos frecuentes en los pacientes descritos. Entre las anomalías oftalmológicas, se incluyen: microcórnea, atrofia óptica, alacrimia con hipoestesia corneal, miosis y coloboma de nervio óptico20.
En relación con el aparente aumento del riesgo tumoral, a la fecha se ha descrito a 4 pacientes con teratoma sacrococcígeo, uno con hepatoblastoma, uno con neoplasia retroperitoneal y otro con ependimoma extradural mixopapilar2-6,20, habiendo remitido a nuestro paciente a Oncohematología Infantil para seguimiento.
Pese a que la esperanza de vida de los pacientes descritos se limita a los primeros años de vida, la mejora en los programas de seguimiento puede estar contribuyendo al aumento de la misma, teniendo nuestro paciente 4 años y medio en la actualidad.
En relación con la base molecular del síndrome, se ha identificado en nuestro paciente una mutación de novo, patogénica, en heterocigosis, en el exón 4 del gen SETBP1. Esta mutación no había sido previamente descrita. A la fecha, las mutaciones en este gen, incluida la de nuestro paciente, se localizan en una estrecha franja de 11 nucleótidos, afectando a 3 de 4 aminoácidos consecutivos, postulándose un mecanismo de ganancia de función o un efecto dominante negativo aun por dilucidar9.
Se trata de una entidad autosómica dominante, siendo todas las mutaciones descritas de novo8-11. El riesgo de recurrencia para los padres en un siguiente embarazo se considera bajo, estimado en un máximo del 1%, al no poder descartar mosaicismo germinal, no aumentando el riesgo de descendencia afectada para el resto de familiares.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos a la familia del paciente su colaboración y consentimiento para compartir sus datos clínicos y la publicación de sus fotografías.
