


Sr. Editor:
Presentamos el caso de una niña marroquí de 7 años que acude a la consulta de Dermatología para reevaluación de un cuadro cutáneo que comienza a los 6 meses de vida mediante placas eritematosas en ambas mejillas y en las zonas fotoexpuestas, sobre las que ocasionalmente se han desarrollado vesículas. Posteriormente estas placas se han ido extendiendo a otras localizaciones entre las que se incluye la superficie extensora de extremidades y glúteos.
Los exámenes de laboratorio practicados, que incluyen hemograma, bioquímica general, factor reumatoide, PCR, antiestreptolisinas (ASLO), inmunoglobulinas, proteinograma, autoanticuerpos (ANA), sedimento urinario, biopsia cutánea, serie ósea y RM cerebral ofrecieron resultados dentro de la normalidad salvo ANA(+) con valores de 1/120, que se han repetido en posteriores determinaciones.
Durante su desarrollo ha presentado un claro retraso ponderoestatural y ha sido revisada por el servicio de oftalmología de nuestro hospital al presentar un astigmatismo miópico congénito, por el cual en la actualidad sigue en revisión. A los 5 años de edad presenta artralgias en la rodilla derecha con normalidad de todos los estudios de imagen desarrollados hasta el momento actual.
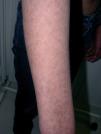
En relación a la sintomatología cutánea, la paciente presenta importante fotosensibilidad, y ha desarrollado quemaduras de primer y segundo grado tras exposiciones solares no superiores a 40 min, por lo que se le ha aconsejado uso de fotoprotectores físicos y medidas de fotoprotección adecuadas. El pelo se ha ido haciendo progresivamente más fino y las uñas presentan onicosquicia y discretas estriaciones longitudinales. Las placas eritematosas previamente descritas han producido con el paso de los años un mosaico de hiper e hipopigmentaciones residuales de aspecto poiquilodérmico (fig. 1).
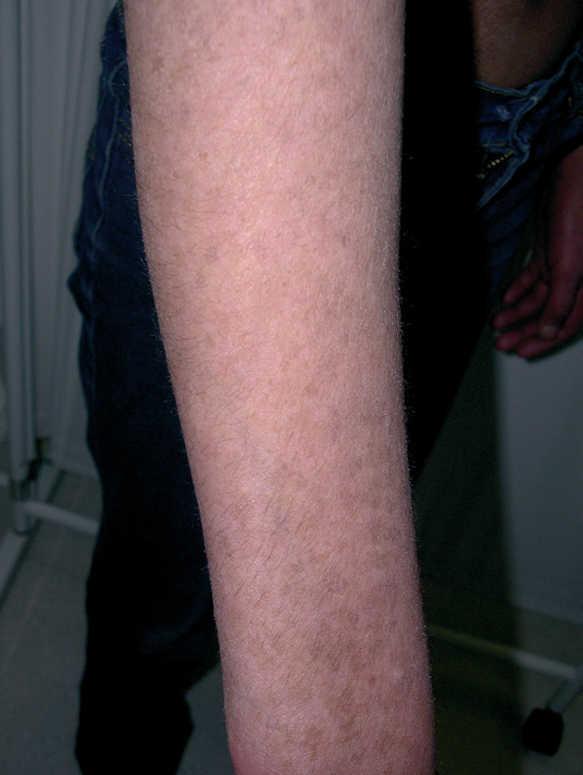
Figura 1. Hipopigmentaciones e hiperpigmentaciones residuales de aspecto poiquilodérmico en la cara dorsal del brazo derecho.
Recientemente su familia ha acudido a revisión con una hermana pequeña (matrimonio consanguíneo marroquí de 5 hijos, 3 hijos y 2 hijas) que presenta a la edad de 3 años lesiones similares a las desarrollada por su hermana con la edad.
Dada la evolución de la paciente, sus antecedentes familiares y la sintomatología reseñada hemos establecido el diagnóstico de síndrome de Rothmund-Thomson.
El síndrome de Rothmund-Thomson es una genodermatosis autosómica recesiva, descrita por el oftalmólogo alemán Rothmund en 1868, caracterizada desde el punto de vista cutáneo por una marcada fotosensibilidad y lesiones de características poiquilodérmicas que parece tener su origen en un mosaicismo genético localizado en el cromosoma 8 1.
La sintomatología comienza a desarrollarse entre el tercer y sexto mes de vida mediante la presencia de placas eritematosas, de aspecto reticulado, a veces con leve hiperqueratosis, que a lo largo de su evolución, dejan hipo e hiperpigmentaciones residuales, lo cual le confiere a la zona afectada su tan característico aspecto poiquilodérmico. En la mayoría de los casos reflejados en la literatura médica las lesiones comienzan en la cara y se extienden a los glúteos y la superficie extensora de las extremidades.
Este síndrome incluye una constelación de manifestaciones clínicas entre las que destacan las siguientes: pelo ralo y fino, encanecimiento prematuro, desarrollo de cataratas juveniles en el 50 % de los pacientes entre los 4 y 7 años de edad, retraso mental, micrognatia, onicodistrofia, malformaciones esqueléticas, hipogonadismo hipogonadotropo y tumores cutáneos asociados a la exposición a radiación ultravioleta.
El mecanismo que subyace al desarrollo de la sintomatología cutánea en estos pacientes parece ser un alteración en los mecanismos de reparación del ADN (la alteración del un gen que codifica la helicasa RECQL4, parece ser responsable de muchos de estos casos) 2. Esto se traduce en una importante inestabilidad cromosómica en la síntesis del ADN en los fibroblastos, acentuada sobre todo tras la exposición a la radiación UVA. En nuestra paciente y su familia no se ha podido realizar estudio genético por motivos técnicos, aunque consideramos interesante su realización, si bien los patrones genéticos del síndrome de Rothmund-Thomson son variados e incluso se han observado pacientes con un cariotipo normal sin ninguna alteración. Es interesante y será objeto de futuros estudios el establecer una correlación entre las alteraciones en el genotipo y su expresión fenotípica.
Es importante realizar un correcto diagnóstico diferencial con otros procesos, tales como el síndrome de Cockayne, la tricotiodistrofia, la disqueratosis congénita, la anemia de Fanconi, el síndrome de Bloom, etc., y en su evolución es conveniente revisiones multidisciplinares anuales dada la cantidad de alteraciones que este síndrome lleva aparejadas 3. En nuestro punto de vista personal creemos que muchos de estos casos, sólo la evolución clínica de los pacientes permite llegar al correcto diagnóstico de éstos, ya que en ocasiones, y como ocurre en el caso presentado, las manifestaciones se solapan con las incluidas en otros procesos de presentación más frecuente como el lupus eritematoso sistémico, diagnóstico atribuido a nuestra paciente durante varios años.
Desde el punto de vista dermatológico es preciso estar atento al posible desarrollo de neoplasias cutáneas, en edades más precoces que en el resto de la población, entre las que ya se han descrito la enfermedad de Bowen, los carcinomas basocelulares, el porocarcinoma ecrino y los carcinomas espinocelulares 4. En otra índole también se han descrito fibrosarcomas, mielodisplasia 5, adenomas de paratiroides, carcinomas gástricos y osteosarcomas asociados. Esto hace imprescindible insistir en las correctas medidas de fotoprotección que debe realizar el paciente desde su infancia y que permite una expectativa de vida similar al del resto de la población. El consejo genético de estos pacientes es aconsejable.
