


La telangiectasia hemorrágica hereditaria (HHT) o síndrome de Rendu-Osler-Weber es una entidad de las consideradas como «enfermedades raras» (afecta a uno de cada 3.000–8.000 individuos) cuya herencia autosómica dominante va determinada por la alteración en la codificación de los genes endoglina (ENG) y activin like Kinase 1, (ALK1) que causan el tipo 1 y 2 respectivamente de la HHT y conducen a displasia en el endotelio de la pared vascular debido a haploinsuficiencia para endoglina. Esto da lugar a una serie de manifestaciones clínicas que consisten básicamente en epistaxis repetidas, telangiectasias mucocutáneas y malformaciones arteriovenosas (MAV) viscerales. A continuación se presenta el caso clínico de un niño de 11 años que desarrolló hipoxemia franca debido a múltiples fístulas arteriovenosas pulmonares.
Hereditary haemorrhagic telangiectasia or Rendu-Osler-Weber syndrome is a rare genetic autosomic dominant disorder with an estimated prevalence of one in 3000–5000 individuals. This multisystemic vascular dysplasia is determined by the mutation of two main genes which are endoglin (ENG) or HHT1 and ALK1 or HHT2. These mutations induce the vascular disorders which cause recurrent epistaxis and eventually multiple telangiectasias and arteriovenous visceral malformations (AVM).
We report the case of an 11-year-old boy who developed severe hypoxaemia due to multiple pulmonary arteriovenous malformations.
El síndrome de Rendu-Osler-Weber, también conocido como telangiectasia hemorrágica hereditaria (HHT), es una enfermedad genética de herencia autosómica dominante cuya prevalencia media es de 1 caso por cada 3.000–8.000 individuos1. Se caracteriza por la gran variabilidad clínica entre distintas familias, e incluso entre pacientes de la misma familia, siendo mayor y más intensa la aparición de síntomas conforme avanza la edad. La clínica viene caracterizada por la presencia de epistaxis espontáneas y recurrentes, la aparición de múltiples telangiectasias mucocutáneas de forma característica en labios, cavidad oral, nariz, punta y alrededor de los dedos, así como por el desarrollo de malformaciones arteriovenosas viscerales, fundamentalmente pulmonares, gastrointestinales, hepáticas y del sistema nervioso central10.
La variante HHT1 se origina por la mutación en el gen endoglina (ENG), mientras que la HHT2 por la alteración del gen ALK1, ambas dando lugar a alteraciones en el endotelio vascular de forma multisistémica4,5.
Caso clínicoEl caso clínico corresponde a un varón de 11 años de edad en cuyos antecedentes familiares destaca su abuela materna con HHT fallecida por hemorragia pulmonar de joven. Su madre presenta epistaxis recurrentes y telangiectasias labiales aisladas. Antecedentes personales de asma episódica intermitente con episodios de broncoespasmos leves en relación a infecciones respiratorias que se controlaban con salbutamol, raramente precisando corticoides orales, y con mejoría significativa en los últimos años. Había consultado por astenia dos meses antes, realizándose analítica que fue normal (Hb 15,4g/dl y Hto 44,8%). Un mes antes del diagnóstico, acude a urgencias a otro hospital por tos en accesos de 10 días de evolución, así como astenia y baja tolerancia al ejercicio. Se realiza Rx tórax, informándose con signos de atrapamiento aéreo. Presenta una saturación de oxígeno por pulsioximetría del 86% que mejora discretamente a 92% con salbutamol inhalado y oxigenoterapia. A pesar de presentar una auscultación normal sin signos de distress respiratorio, se diagnostica de «crisis asmática leve» e infección respiratoria y se remite a domicilio con salbutamol y budesonida inhaladas más metilprednisolona y claritromicina vía oral.
Acude a control a su centro de salud varios días después. La auscultación pulmonar es normal sin signos de atrapamiento aéreo ni distress respiratorio, objetivándose una saturación del 88%. Se realiza ECG presentando ritmo sinusal con signos sugerentes de crecimiento ventricular izquierdo. TA normal y glucemia normal Se remite a urgencias para estudio por hipoxemia de probable etiología cardiaca.
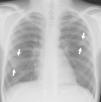
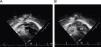
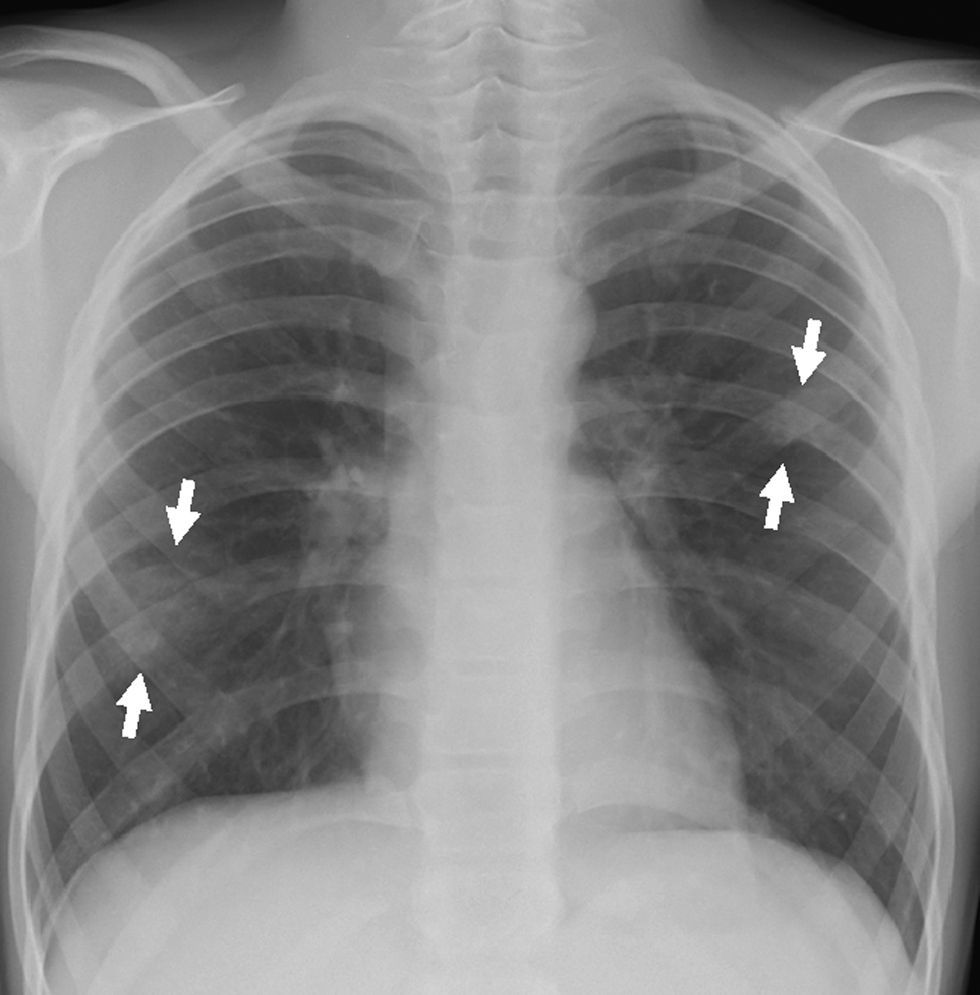
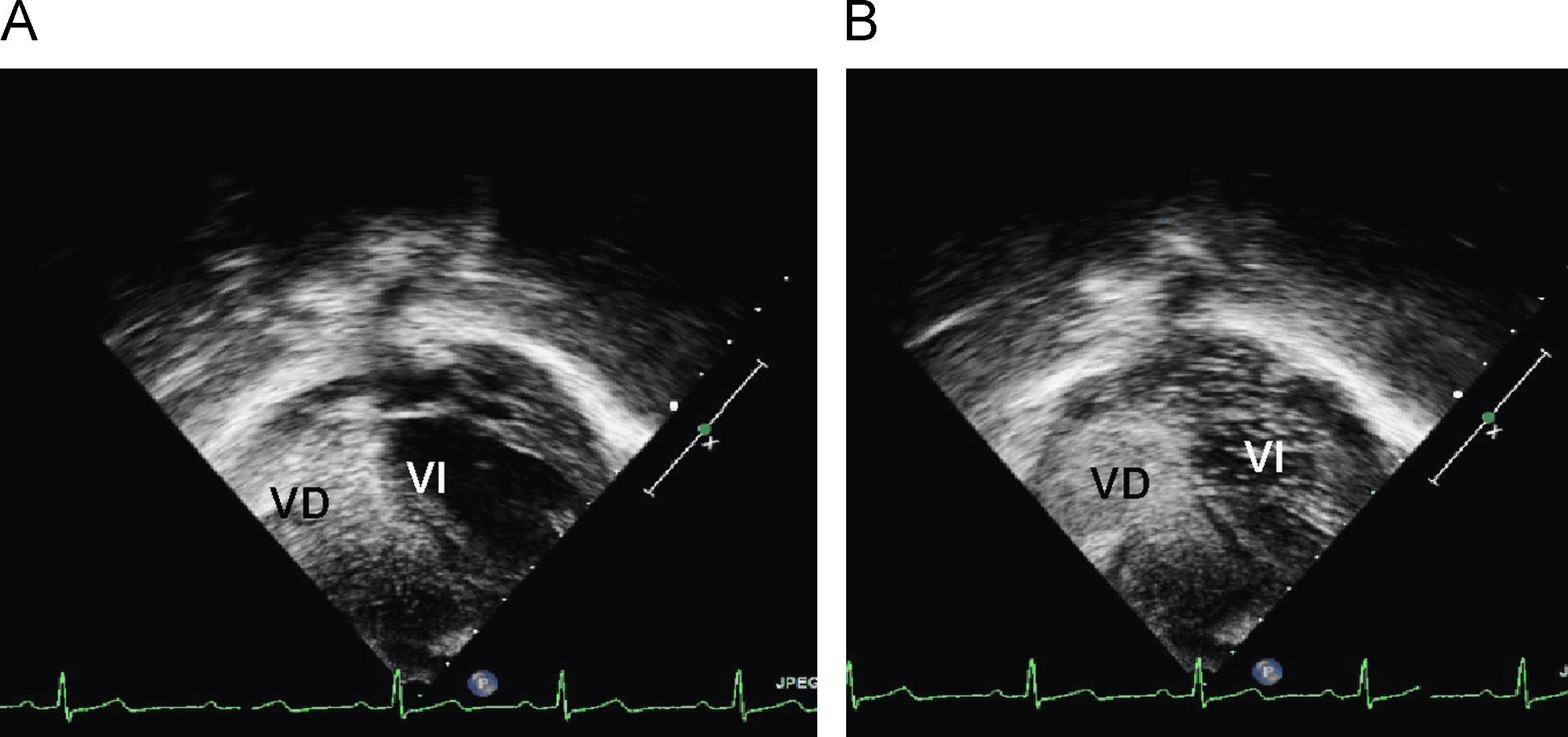
En urgencias se constata ventilación pulmonar normal, ausencia de cianosis, deformidades ungueales leves, Rx tórax normal y ausencia de mejoría con la administración de oxígeno. La analítica muestra discreta policitemia (Hb 16,3g/dl y Hto 48,9%), bioquímica normal, ionograma normal, pH 7,37 con pCO2 46mmHg y pO2 29,6mmHg (capilar). Se consulta al cardiólogo quien realiza un ecocardiograma que muestra función ventricular normal, sin objetivarse alteraciones estructurales, ni drenajes venosos sistémicos a aurícula izquierda. Se plantea la posibilidad de fístulas arteriovenosas pulmonares una vez excluidas otras posibilidades de cortocircuito derecha-izquierda. Se reinterroga a la madre sobre antecedentes familiares de fístulas AV pulmonares o de enfermedad de Rendú Osler. Se reexamina la radiografía de tórax en busca de lesiones sugerentes de fístulas (fig. 1) y se realiza ecocardiograma con inyección de suero salino según protocolo de estudio de fístulas arteriovenosas pulmonares. Tras inyección periférica de suero salino agitado se observa relleno de cavidades derechas y reaparición tardía (tras 4 latidos) en cavidades izquierdas (fig. 2).
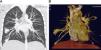
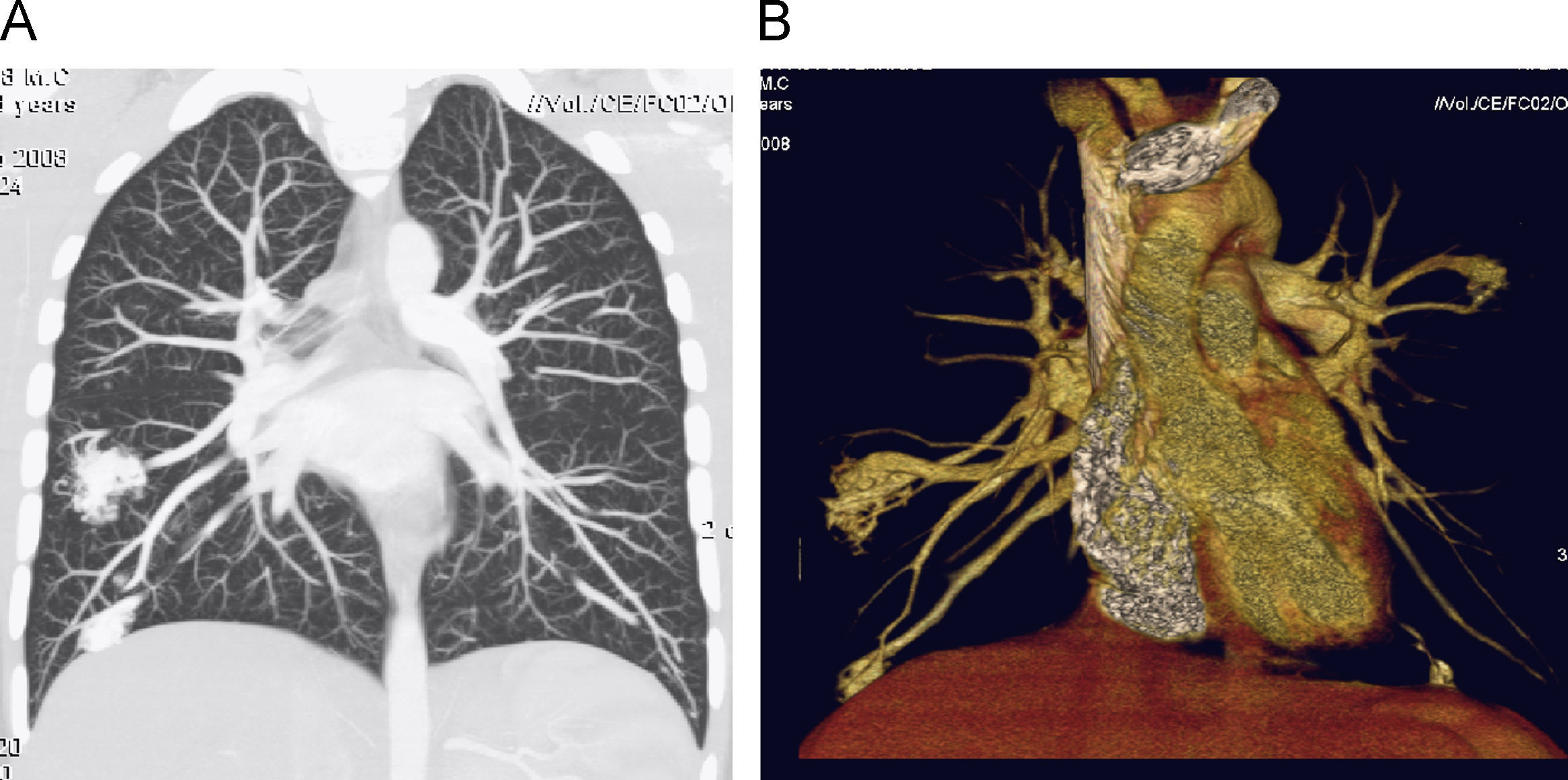
Se ingresa durante 24h para completar estudio de enfermedad de Rendú-Osler con los siguientes resultados: angioTC torácico con múltiples fistulas AV pulmonares (fig. 3), TC abdominal con imágenes sugerentes de pequeñas telangiectasis hepáticas, estudio de sangre oculta en heces negativo, angioresonancia cerebral con imagen sugerente de pequeña malformación AV. El estudio genético ha demostrado una mutación V416fs con una delección de 13 nucleótidos en posición c1248_1260 en el exón 9a de la endoglina. Es dado de alta con oxígeno a domicilio intermitente para mantener saturaciones mínimas del 90% y tratamiento con amlodipino.
Se realiza cateterismo derecho e izquierdo, programado, lográndose embolización percutánea de dos fístulas AV pulmonares grandes con dispositivos de Amplatzer ductal en lóbulos medio e inferior derechos. La evolución es favorable tras el cateterismo, con ascenso de la saturación de oxígeno a 90–92% y mejor tolerancia al ejercicio físico. Se repite ecocardiograma de control que muestra persistencia del relleno de cavidades izquierdas, subjetivamente en menor cantidad que en el estudio inicial. En la actualidad se encuentra pendiente de valorarse nuevas embolizaciones.
DiscusiónLa HHT, descrita por primera vez por Sutton en 1864 y más ampliamente por Rendu en 1896, siendo Osler en 1901 y Weber en 1907 quienes publican las primeras series de casos, es una enfermedad autosómica dominante considerada hasta ahora como «enfermedad rara». Sin embargo, datos publicados más recientemente indican que en realidad no sea «tan rara», con prevalencias en Cantabria de hasta 8,2 por cada 10.000 habitantes1. En el momento actual existen grupos de trabajo distribuidos por todo el mundo que establecen protocolos de diagnóstico y tratamiento para estos pacientes2. En nuestro país, el Hospital de Sierrallana de Torrelavega con el grupo de trabajo dirigido por el Dr. Roberto Zarrabeitia, está reconocido como centro de referencia en nuestro país por la HHT Foundation International (Monkton. MD. EE.UU.) desde el año 2003, habiéndose celebrado en el 2009 el 8.° Congreso Mundial de HHT en Santander3.
Reconocida la heterogeneidad génica en la etiopatogenia de la HHT, hoy día se reconocen dos genes principales cuya mutación da lugar a la enfermedad: el gen de la endoglina (ENG) situado en el 9q3,3–q3,4 (variante HHT1) y el gen ALK situado en 12q13 (variante HHT2)4–6. Ambos genes intervienen en la angiogenesis codificando proteínas que atraviesan la membrana de las células endoteliales formando parte del sistema señalizador TGF-beta, citoquina transmisora de señales entre el exterior y el interior del endotelio. La endoglina cumple además una función estructural en la propia célula endotelial7. Por diversos mecanismos, actualmente todavía en estudio, la mutación de estos genes determina las telangiectasias (dilataciones de vénulas postcapilares) y malformaciones arteriovenosas que caracterizan a la HHT8. Además, trabajos recientes9 sugieren que en un 1–3% de los casos se detecta una mutación en el gen SMAD4. Esta última mutación se asocia a poliposis intestinal juvenil, por lo que estos pacientes tienen riesgo de neoplasia gastrointestinal y deben ser estrechamente controlados en este sentido.
Los criterios diagnósticos, definidos por un comité de expertos en 1999 en la isla de Curaçao (conocidos como criterios de Curaçao)10 son cuatro:
- 1.
Epistaxis espontáneas y recurrentes.
- 2.
Telangiectasias mucocutáneas características.
- 3.
Malformaciones arteriovenosas viscerales.
- 4.
Historia familiar entre parientes de primer grado.
El diagnóstico es definitivo si se cumplen tres o más criterios, probable si se cumplen dos y poco probable si se cumplen menos de dos.
Las epistaxis suelen ser espontáneas y recurrentes y es el síntoma más frecuente y por el que suele debutar la enfermedad: el 46% a los 10 años, el 70% a los 20 años y el 100% a los 40 años11. Según avanza la edad suelen aumentar en frecuencia e intensidad. Existe tal variabilidad que se dan desde esporádicas y de escasa cuantía hasta importantes que requieren múltiples transfusiones. En ocasiones, son tan frecuentes, que pueden afectar significativamente la vida de los pacientes.
Las telangiectasias mucocutáneas suelen debutar entre 5 y 20 años tras las epistaxis, aumentando con la edad, y suelen localizarse en palmas de las manos, lecho de las uñas, labios, lengua y cara. Raramente presentan sangrado de escasa cuantía. En pediatría, las telangiectasias se pueden incluir como criterio diagnóstico si se observan a nivel de capilaroscopia2.
De las malformaciones AV viscerales, las más frecuentes son las pulmonares (MAVP) que presentan hasta un 50% de los pacientes12. El 70% de las fístulas pulmonares se presentan en pacientes con HHT. Pueden aparecer a cualquier edad, aumentando en número y tamaño con el tiempo. Clínicamente dan lugar a hipoxemia por cortocircuito derecha-izquierda a nivel pulmonar con grados variables de disnea, cianosis y policitemia. Pueden originar embolismos a la circulación cerebral con abscesos y accidentes isquémicos cerebrales y hemorragias por rotura de las fístulas, siendo estas raras, pero de mayor riesgo durante el embarazo. El diagnóstico se realiza mediante ecografía transtorácica con contraste, utilizando suero salino agitado para producir múltiples burbujas de aire que se inyecta en una vena periférica, siendo positiva la prueba si aparecen al cabo de 3 a 5 latidos en cavidades izquierdas13,14. La arteriografía pulmonar confirma su tamaño y localización. Hoy día también se dispone de angioTC de alta resolución que se utiliza no solo con fines diagnósticos, sino también para guiar la embolización terapeútica, así como los controles postembolización.
Aunque el 80% de los pacientes con HHT tienen telangiectasias gastrointestinales de muy diverso tamaño y cuantía, demostradas por endoscopia en estudios recientes15, especialmente gástricas y duodenales, solo presentan sangrado sintomático un 25–30% de los pacientes, normalmente hacia la 5.a–6.a década de la vida. Son más frecuentes en mujeres y dan lugar a anemia ferropénica por sangrado crónico más que a episodios de hemorragia digestiva aguda.
Las malformaciones vasculares hepáticas, antes consideradas menos frecuentes por su escasa y poco frecuente sintomatología, han pasado a ser diagnosticadas hasta en un 70% de los casos debido a las técnicas de TC helicoidal con contraste16. Raramente producen insuficiencia cardiaca por shunt, síntomas de hipertensión portal o de afectación biliar.
Por último, las malformaciones vasculares del SNC se dan en aproximadamente el 23% de los casos y pueden estar presentes desde el nacimiento. Están descritas desde malformaciones arteriovenosas hasta telangiectasias y aneurismas, pudiendo coexistir en el mismo paciente17. Pueden dar lugar a cefaleas, epilepsia y accidentes cerebrovasculares isquémicos o hemorrágicos. Ciertos autores describen un riesgo de hemorragia cerebral de hasta 20 veces mayor en menores de 45 años que en la población general18.
En cuanto al tratamiento, consiste fundamentalmente en controlar el sangrado oral y nasal, así como prevenir las complicaciones hemorrágicas y limitar la posibilidad de accidentes cerebrovasculares secundarios a embolismos sistémicos de las fístulas AV viscerales. Aquí, se incluye la profilaxis antibiótica ante cualquier tipo de procedimiento con riesgo bacteriémico. En el control de las epistaxis, además de la limpieza con solución salina y el uso diario de lubricantes nasales, se utilizan métodos como la dermoplastia septal, ablación local con láser y tratamientos locales y sistémicos hormonales (de ellos, el raloxifeno ofrece resultados esperanzadores) y con antifibrinolíticos como el ácido tranexámico con buenos resultados19. Otras drogas en estudio con resultados positivos son los agentes antiangiogénicos (bevacizumab), el interferón, los inmunosupresores (sirolimus y tacrolimus) y la n-acetilcisteína19.
Las MAVP mayores de 3mm deben embolizarse con diversos tipos de dispositivos, las pequeñas pueden embolizarse con espirales y las grandes con dispositivos tipo Amplatzer20, consiguiendose al menos temporalmente buenos resultados, ya que algunas pueden recanalizarse y pueden aparecer otras nuevas.
Otras malformaciones AV viscerales pueden ser susceptibles de tratamiento quirúrgico o de embolización según la sintomatología. Como medidas generales puede ser necesaria la ferroterapia oral y/o las transfusiones según el grado de anemia.
Como comentarios finales, queremos destacar que, aunque la hipoxemia de causa respiratoria es con mucho la más frecuente en la infancia, en ausencia de clínica respiratoria la hipoxemia franca nos debe hacer valorar otros diagnósticos más «raros» como el que nos ocupa. La desaturación leve y progresiva con frecuencia no causa cianosis y solo se hace aparente a medida que se hace más intensa o que se asocie a policitemia. En estos casos es necesario realizar una correcta anamnesis buscando antecedentes familiares, ya que este tipo de diagnósticos pueden pasar desapercibidos y debutar con complicaciones hemorrágicas o embólicas en la edad adulta. La ausencia de telangiectasias faciales y/o de epistaxis repetidas no excluye la posible existencia de fístulas pulmonares, como el caso que nos ocupa, aunque estos hallazgos nos deben poner sobre aviso de esta enfermedad. Se deben hacer estudios genéticos en unidades de referencia que permitan hacer un correcto diagnóstico y un screening a los familiares de primer grado de los casos positivos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr. Roberto Zarrabeitia de la Unidad de HHT del Hospital de Sierrallana por su colaboración y revisión de nuestro trabajo.
