



El HGPS (Hutchinson-Gilford progeria syndrome ‘síndrome de progeria de Hutchinson-Gilford’) es una enfermedad rara, de herencia autosómica dominante, con una incidencia estimada de 1:4 millones de nacidos vivos1, y descrita por Jonathan Hutchinson en 18862. En 1904, Hastings Gilford3 acuñó el término progeria, derivado del griego pro=temprano y geras=viejo.
En la mayoría de los casos, las manifestaciones clínicas son evidentes después del primer año, con una edad promedio al momento del diagnóstico de 2,9 años4. La apariencia al nacer es normal; la disminución importante del crecimiento ocurre durante el primer año. Presentan dismorfismo craniofacial con ojos prominentes, nariz ganchuda, labios delgados, dentición anormal, micrognatia, orejas prominentes con ausencia de lóbulos auriculares, alopecia, venas cranianas prominentes, pérdida de grasa subcutánea, rigidez articular, cambios óseos y cambios cutáneos, que son aparentes durante el segundo al tercer año de vida. La muerte suele ocurrir en la adolescencia, secundaria a complicaciones cardiovasculares. En 2003, Ericksson et al5 identificaron mutaciones en el gen lamina A en 20 de 23 pacientes con HGPS. De Sandre-Giovannoli et al6 identificaron mutaciones en el mismo gen en 2 pacientes.
Se presenta un paciente masculino de 2 años de edad, de raza mestiza y procedencia urbana, enviado a endocrinología pediátrica por talla baja, alopecia y piel seca.
Producto de un segundo embarazo complicado por amenaza de aborto en el primer trimestre y polihidramnios sin alteraciones morfológicas fetales en el segundo trimestre. El parto fue por cesárea a las 39 semanas de edad gestacional. El peso y la talla al nacer eran de 3.400g y 52cm, respectivamente. La puntuación en el test de Apgar fue de 7/7 y el examen físico resultó normal.
A los pocos dias de nacido se observó piel acartonada, seca, con lesiones en el tronco y las extremidades y, a partir de los 2 meses, retraso de crecimiento (peso y talla) e hipotonía leve.
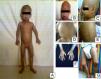
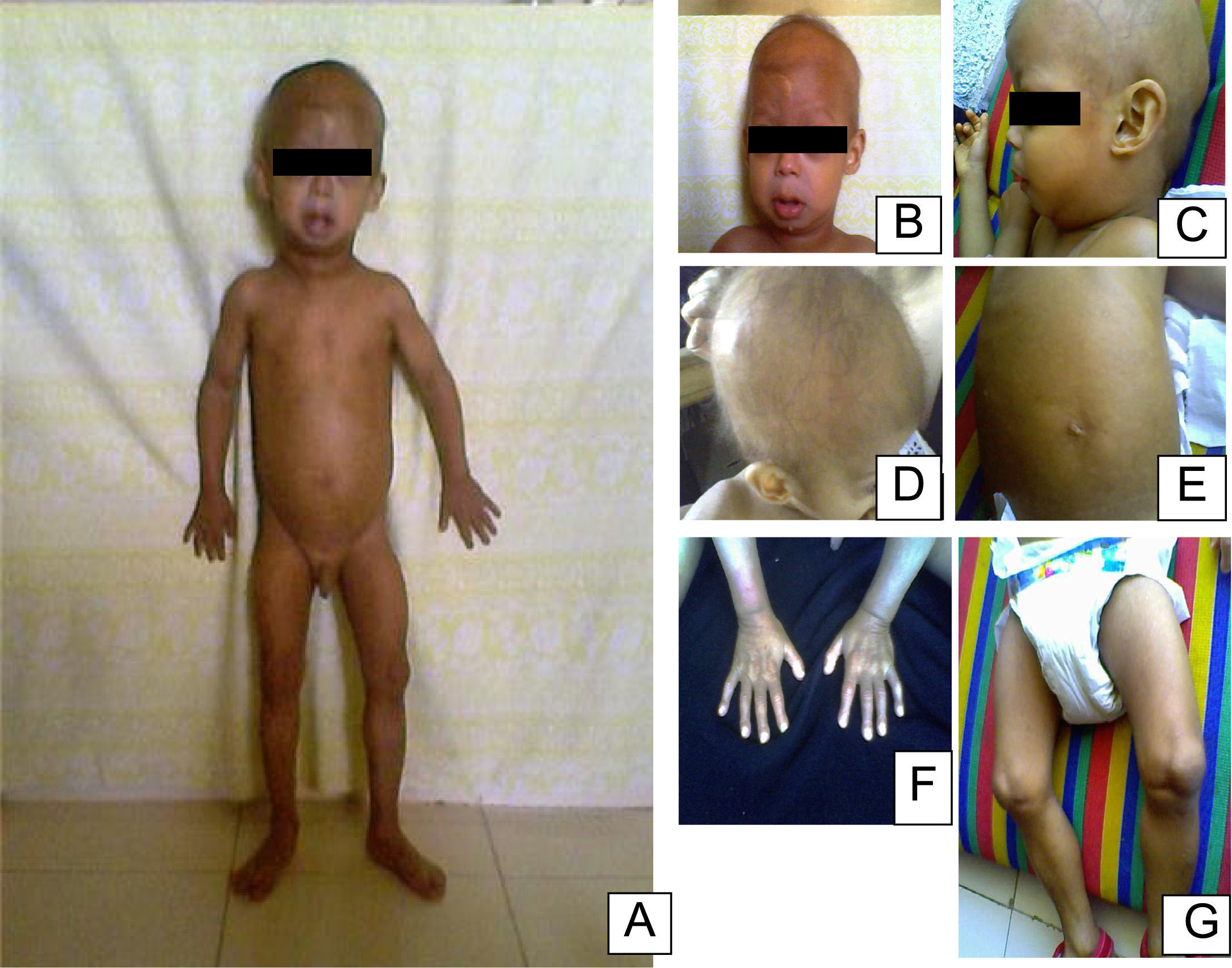
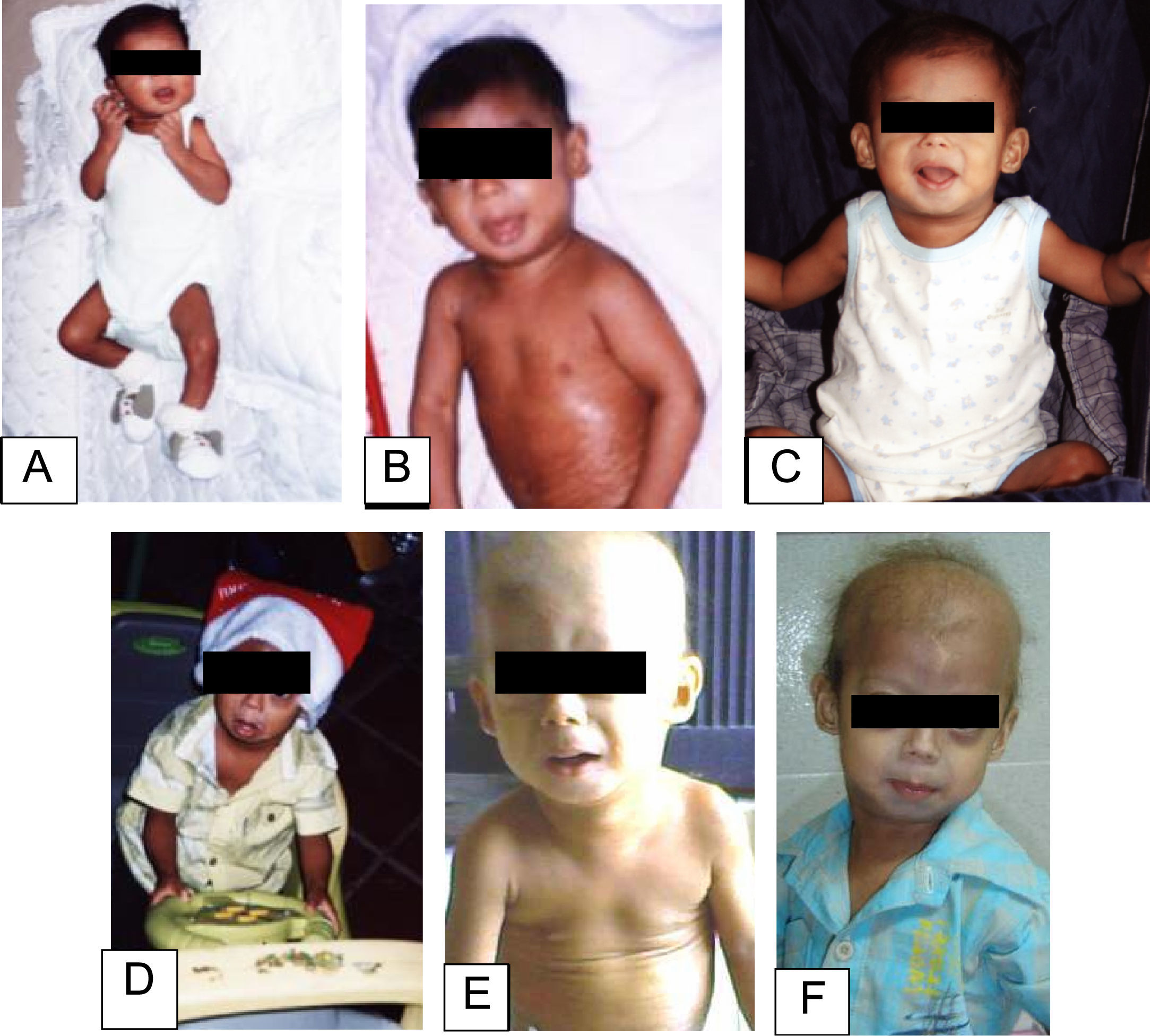
En el examen físico a los 2 años se observaron las siguientes características: peso de 8kg (<3%), talla de 77cm (<3%) y un índice de masa corporal inferior al 3%. Las características craniofaciales eran cabeza desproporcionada, micrognatia, venas del cuero cabelludo prominentes, alopecia generalizada, ojos prominentes, pestañas y cejas escasas, punta de nariz picuda y esculpida, labios finos, orejas prominentes, ausencia de lóbulos auriculares, voz aguda, retraso en la dentición. El tórax era piriforme; la exploración mostró ruidos cardíacos rítmicos sin soplos y buena entrada de aire en ambos campos pulmonares. La piel era delgada, tirante, seca, arrugada, con lesiones hiperpigmentadas y tenía pérdida de grasa subcutánea. Las extremidades eran delgadas con postura de montar a caballo y rigidez articular (fig. 1). El examen neurológico y de desarrollo psicomotor resultaron normales. La progresión del fenotipo es evidente en la figura 2. En los estudios de laboratorio, se realizó una biopsia de piel con evidencia de esclerodermia. El resto de los estudios fueron normales (biopsia del duodeno, endoscopia digestiva, perfil tiroideo, electrolitos séricos, pruebas de función renal, cariotipo, ecocardiograma, ecografía abdominal, radiografía de tórax, glucemia y carpograma). Se llegó al diagnóstico clínico de progeria de Hutchinson-Gilford. Se realizó confirmación molecular al hallar una mutación definida como c.1824 C>T (p. Gly608Gly) en el exón 11 del gen LMNA.
El HGPS pertenece a un grupo de trastornos asociados a mutaciones en el gen LMNA o «laminopatías», entre los que se incluyen distrofia muscular de Emery-Dreyfuss, miocardiopatía dilatada, enfermedad de Charcot-Marie-Tooth tipo 2B, distrofia muscular de cintura tipo 1B y lipodistrofia familiar parcial tipo Dunnigan.
De acuerdo con nuestro conocimiento, éste es el primer colombiano reportado en la literatura médica con HGPS. Los estudios moleculares detectaron la mutación encontrada con más frecuencia en los pacientes con HGPS, un cambio de C>T en el nucleótido 1824 (p. Gly608Gly) en el exón 11 del gen LMNA, que activa un sitio de empalme críptico, lo que se traduce en la producción de una proteína que suprime 50 aminoácidos cerca del carboxi-terminal (progerina). Esta proteína anormal es farnesilada y permanece unida a la membrana nuclear interna, distorsionándola e interrumpiendo su función. El descubrimiento del mecanismo molecular ha permitido el uso experimental de drogas que inhiben la enzima farnesiltransferasa en el tratamiento del HGPS con resultados prometedores en modelos animales. Nuestro paciente será incluido en un ensayo clínico con utilización de inhibidores de la farnesiltransferasa.
Se ha demostrado que la progerina se acumula en células humanas al envejecer7. Las preguntas que nos hacemos ahora son: ¿cuánto nos enseñará la genética de la progeria de Hutchinson-Gilford acerca del mecanismo del envejecimiento normal? ¿Son los inhibidores de la farnesiltransferasa la fuente de la eterna juventud?
