



El síndrome de Peutz-Jeghers es un raro proceso hereditario que suele iniciarse en la infancia. Se caracteriza por la presencia de lesiones cutáneas pigmentadas y pólipos gastrointestinales. Numerosos estudios revelan una incidencia elevada de cáncer (gastrointestinal y extradigestivo) en estos enfermos y su aparición a temprana edad, así como su asociación con tumores ováricos y testiculares. Por ello, es necesario un estrecho seguimiento y un tratamiento agresivo de estos enfermos.
Presentamos 2 hermanos afectados de síndrome de Peutz-Jeghers cuyo padre y abuelo fallecieron a consecuencia de cáncer digestivo relacionado con la enfermedad.
Peutz-Jeghers syndrome is an inherited disorder which usually debuts during childhood. It is characterized by mucocutaneous pigmentation and hamartomatous polyps in the gastrointestinal tract. Numerous reports indicate a high incidence of gastrointestinal and extraintestinal cancer in these patients, their appearance at a young age, as well as its association with ovarian and testicular tumors. An aggresive approach of these patients seems to be necesary.
We report the case of two brothers suffering from Peutz-Jeghers syndrome whose father and grandfather died as a consecuence of the progression of an intestinal cancer related to the syndrome.
El síndrome de Peutz-Jeghers (SPJ) es un trastorno poco frecuente, de herencia autosómica dominante, caracterizado por la asociación de pigmentación cutáneo-mucosa y poliposis intestinal. El primero en describir el síndrome fue Peutz en 1921, y fue Jeghers, unos años más tarde, quien realizó la descripción definitiva.
A pesar de ser una condición hereditaria, sólo en la mitad de los casos se encuentran antecedentes familiares. Su incidencia aproximada es de 1 por cada 8.300-29.000 recién nacidos1. En la mayoría de los pacientes los primeros síntomas aparecen durante la infancia. Se ha descrito en todas las razas y todos los tipos de piel2.
Presentamos el caso de dos hermanos diagnosticados de SPJ que nacen en el seno de una familia con otros miembros afectados por la enfermedad.
CASO CLÍNICONuestro primer paciente es un varón que consultó por primera vez a los 9 años de edad, por dolor abdominal recurrente con vómitos ocasionales. Su padre había sido visto en nuestro hospital a los 32 años por dolor abdominal y rectorragia, y en su exploración destacaba la presencia de melanosis perioral. Se le realizó un estudio endoscópico en el que se encontraron múltiples pólipos en recto y sigma que fueron biopsiados con el resultado de pólipos hamartomatosos y un adenocarcinoma velloso. Presentaba, además, metástasis pulmonares y hepáticas. Falleció 1 año más tarde, pese al tratamiento.
A su vez, el abuelo paterno había fallecido a los 66 años de edad a causa de adenocarcinoma de recto y sigma.
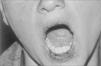
La exploración física del niño fue normal, salvo por la presencia de melanosis perioral (fig. 1). Los análisis de sangre fueron normales y el estudio de sangre en heces, negativo. Se realizó estudio endoscópico en el que se encontró una lesión polipoidea en sigma, que fue extirpada y biopsiada con resultado de pólipo hiperplásico benigno. Con estos hallazgos y los antecedentes familiares el niño fue diagnosticado de síndrome de Peutz-Jeghers, y se le realizó un estrecho seguimiento, clínico y endoscópico.
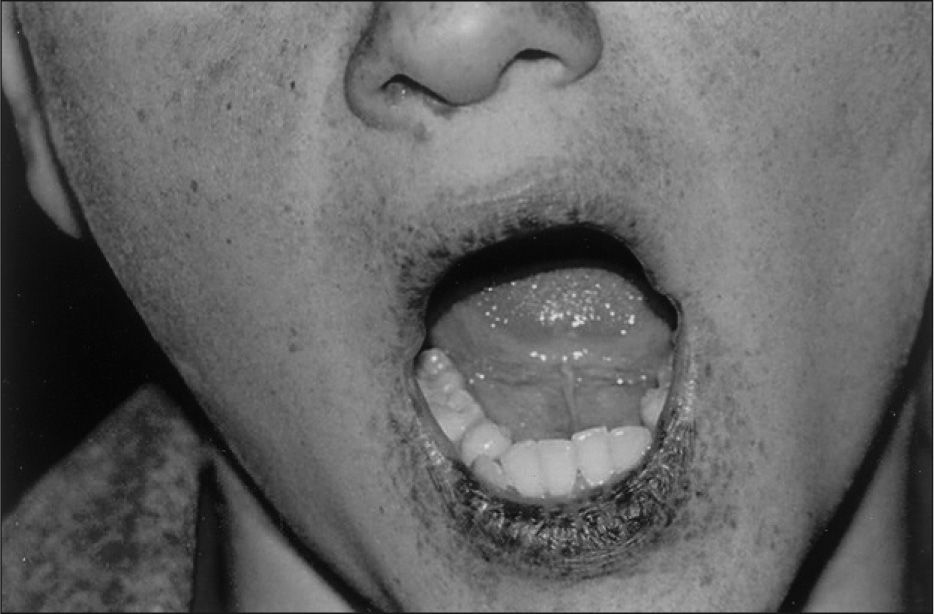
Tras 10 años de seguimiento, el paciente sólo ha presentado cuadros de dolor abdominal esporádico, y ha mantenido cifras normales de hemoglobina, sin pérdida de sangre por heces. Se han realizado tres estudios endoscópicos más; el primero fue normal, pero los dos últimos mostraron la presencia de pólipos pequeños en el colon y duodeno, que fueron extirpados y biopsiados; revelaron la misma histología que el primero.
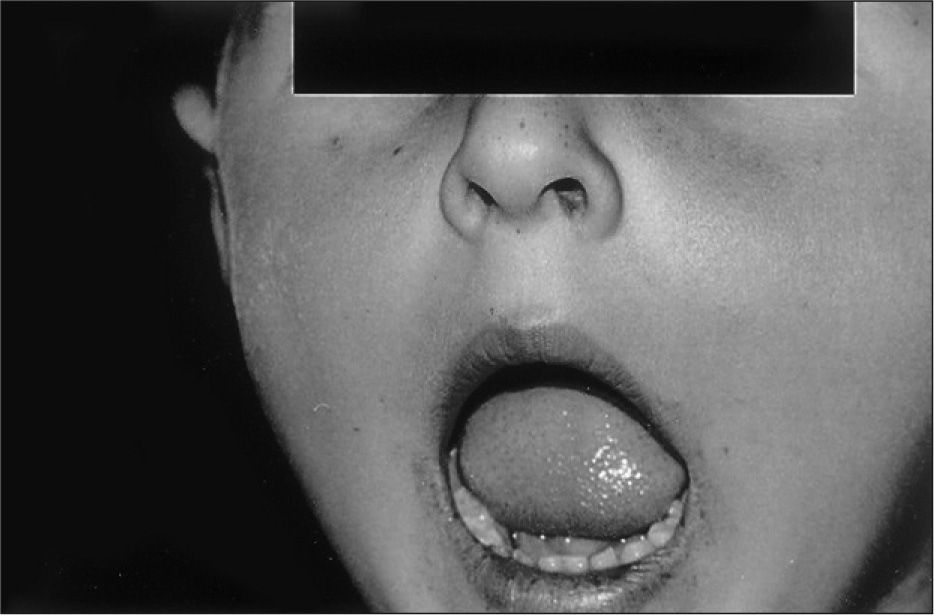
El segundo paciente, hermano del anterior, fue diagnosticado de SPJ a los 2 años por la presencia de melanosis perioral (fig. 2) y los antecedentes familiares. Se realizó un primer estudio endoscópico a la edad de 5 años. Se encontró y resecó un pólipo gástrico (pólipo hiperplásico benigno). A los 7 años presentó una invaginación yeyuno-yeyunal que precisó resección intestinal. A los 12 años se le practicó una nueva endoscopia, en la que se le encontró y resecó dos pólipos gástricos, con igual histología que el previo.
A los 9 años consultó por agrandamiento mamario bilateral, acompañado de aceleración de la velocidad de crecimiento. En la exploración física se vio ginecomastia bilateral con galactorrea y un teste izquierdo de 5ml, de consistencia homogénea. El estudio endocrinológico fue compatible con una pubertad precoz periférica, con adelanto de 3 años en la edad ósea. En la ecografía testicular se observaron imágenes de microlitiasis testicular bilateral. El estudio anatomopatológico del teste izquierdo concluyó la presencia de un tumor multifocal de los cordones sexuales con estructuras anulares. Este testículo fue extirpado, pero se respetó el derecho. La ginecomastia regresó y en los 4 años siguientes no se han vuelto a objetivar anomalías endocrinológicas, el teste derecho es normal en tamaño y consistencia y no presenta nuevas alteraciones ecográficas.
DISCUSIÓNDesde la descripción inicial del síndrome de Peutz-Jeghers, el espectro de manifestaciones clínicas que se incluyen en el mismo se ha ido incrementando con el tiempo.
Las lesiones cutáneas consisten en lunares de color negro-azulado o marrón que suelen localizarse en labios, mucosa oral y zonas periorificiales2,3. No suelen estar presentes al nacer, pero aparecen precozmente, en los primeros 2 años de vida4. No se ha descrito su transformación maligna.
Los pólipos gastrointestinales (hamartomas) se localizan fundamentalmente en el intestino delgado, y con menos frecuencia en el grueso y en el estómago2,3.
En la mayoría de los pacientes los primeros síntomas y muchas de las complicaciones quirúrgicas de la enfermedad aparecen durante la infancia5, y puede ser aparente incluso en el período neonatal6. Todos los casos conocidos han desarrollado el fenotipo completo a los 25 años4.
El motivo de la consulta más frecuente es el dolor abdominal y el sangrado digestivo2. La invaginación representa la principal complicación de la poliposis y es causa de obstrucción intestinal intermitente. En algunos pacientes la primera manifestación es el prolapso o extrusión anal de los pólipos, que, cuando sucede en el primer año de vida, obliga a descartar SPJ, incluso cuando no haya pigmentación5. Hasta el 5,5% de los pacientes presenta pólipos extraintestinales5.
Un pequeño porcentaje de pacientes debuta con síntomas endocrinológicos relacionados con la presencia de tumores gonadales5.
Existe una gran variación interindividual en la intensidad de los síntomas. Esta disociación puede deberse a la distinta edad a la que se realiza el diagnóstico, ya que la aparición de la clínica tiende a ser específica de la edad. Asimismo, se ha documentado variación fenotípica entre familias, y dentro de la misma familia4. Lo que parece no ofrecer dudas es la no benignidad del SPJ, pues la supervivencia suele estar disminuida por la gran cantidad de complicaciones gastrointestinales que implica, que pueden conducir a la muerte incluso en el primer año de vida6, y por el desarrollo precoz de cáncer. Además, los síntomas tienden a ser cada vez más precoces en generaciones sucesivas, y algunos autores7 sugieren que la edad a la que aparecen los tumores también tiende a adelantarse.
Las primeras descripciones de cáncer intestinal con metástasis comienzan en 19658. Se han descrito gran variedad de tumores gastrointestinales y extraintestinales, benignos y malignos: mama, cérvix, testículo, ovario, vesícula biliar y páncreas9–11.
En 1970, Scully12 describe por primera vez el tumor ovárico de cordones sexuales con túbulos anulares (13 casos). Posteriormente. se han descrito más de 100 casos, casi la mitad en enfermas con SPJ. Al contrario que en la población general, en que suele ser unilateral y grande, en estas enfermas suelen ser bilaterales, multifocales y de pequeño tamaño. Estos tumores se han encontrado en casi todas las mujeres con SPJ en las que se ha examinado el ovario, y algunos autores13, los consideran una característica fenotípica del síndrome y no una complicación.
En varones pueden aparecer tumores testiculares de células de Sertoli, que, como en nuestro segundo paciente, suelen manifestarse en la primera década de la vida por ginecomastia, aceleración de la velocidad de crecimiento y adelanto de la edad ósea. Su comportamiento tiende a ser benigno, mientras que en el resto de la población el 10–20 % tienen un curso maligno2.
Recientemente, dos grupos de investigadores han identificado un gen cuyas mutaciones eran responsables de al menos algunos casos de SPJ14,15. Se localiza en el brazo corto del cromosoma 19, codifica una proteína con gran parecido a la serina-treonina-quinasa y ha sido llamado STK11. Posteriormente, Gruber et al16 han aportado evidencia de que este gen actúa como un gen supresor de tumores y podría estar involucrado en las fases tempranas de la transformación de hamartomas en adenocarcinomas, lo que explicaría en parte el elevado riesgo de malignización que presentan estos pacientes.
Con todos estos datos parece incuestionable la necesidad de realizar un estrecho seguimiento de estos enfermos. Cada vez son más los autores que abogan por un tratamiento agresivo2,17 y recomiendan:
- –
Endoscopia digestiva alta y baja cada 2 años, comenzando a los 10 años.
- –
Exploración testicular anual desde los 10 años.
- –
Exploración de mamas anual con mamografía cada 2–3 años desde los 25 años.
- –
Examen pélvico ginecológico en la mujer con ecografía anual desde los 20 años.
- –
Ecografía abdominal y pancreática cada 1–2 años desde los 30 años.
No existe acuerdo en cuanto a la actitud que tomar con los pólipos gastrointestinales. Para algunos autores9, todos deben ser extirpados, mientras que otros creen que sólo es necesario eliminar los mayores de 10 mm3,18, pues es a partir de ese tamaño cuando aumenta claramente el riesgo de malignización.
En cuanto a los tumores testiculares, Gruber et al16 establecen la siguiente pauta: en los tumores unilaterales debe realizarse orquiectomía; en los bilaterales, esta debe reservarse para los casos con mal pronóstico de talla y en los que la biopsia indica que el paciente va a ser estéril. En los demás casos puede realizarse mastectomía y tratamiento con inhibidores de los receptores estrogénicos.
En resumen, son numerosos los estudios que confirman la elevada incidencia de cáncer en estos enfermos, y su aparición a temprana edad. Además, la vida de estos enfermos se ve comprometida por numerosas complicaciones quirúrgicas derivadas de los pólipos. Por todo ello, apostamos por un abordaje agresivo de estos pacientes, e incluso por la extirpación de todos los pólipos, con independencia de su tamaño.
