





El síndrome de Noonan, caracterizado generalmente por talla baja, dismorfia facial, defectos cardíacos y criptorquidia en varones, es una enfermedad autosómica dominante, genéticamente heterogénea. Su origen se encuentra en el 50 % en mutaciones en el gen PTPN11, que codifica la proteína tirosinfosfatasa (SHP2) y da lugar a un aumento de su función y en el 5% a mutaciones del gen KRAS. Recientemente, se ha identificado una nueva mutación en el gen SOS1, que se relaciona aproximadamente con el 20 % de los síndromes de Noonan sin mutación en el gen PTPN11. Esta diferencia en el genotipo produce diferencias fenotípicas que debemos conocer. Presentamos un caso de síndrome de Noonan por una mutación en el gen SOS1 en el que se describe su fenotipo y evolución a lo largo de la infancia y la pubertad.
Noonan syndrome, characterized by short stature, facial anomalies, heart disease and cryptorchidism in males, is an autosomal dominant, genetically heterogeneous disease. Approximately 50 % of Noonan syndrome cases are caused by gain-of-function mutations in PTPN11, encoding the tyrosine phosphatase (SHP2) and 5% are caused by KRAS mutations. Recently, a new mutation in SOS1 gene has been identified in approximately 20 % of cases of Noonan syndrome without PTPN11 mutation. That difference in genotype has a relationship with phenotype that we must investigate. We report a case of Noonan syndrome due to an SOS1 mutation; we describe his phenotype and subsequent outcome.
El síndrome de Noonan, presente en 1 de cada 1.000-2.500 recién nacidos, fue descrito por primera vez en 1968 por Noonan1 y Noonan y Ehmke1,2, aunque la primera descripción fenotípica concordante data de 1883 por Kobilinsky.
Este síndrome, de herencia autosómica dominante, se caracteriza por talla baja proporcionada, rasgos faciales característicos como hipertelorismo, fisuras palpebrales antimongoloides, orejas rotadas y pelo con implantación baja, cubitus valgus, defectos cardíacos congénitos (sobre todo estenosis valvular pulmonar y miocardiopatía hipertrófica), deformidades torácicas y vertebrales, retraso mental leve, alteraciones hematológicas, retraso puberal y criptorquidia en varones. El diagnóstico es fundamentalmente clínico, para lo que nos son útiles los criterios propuestos por Van der Burgt et al3.
Aproximadamente, el 50% de los casos son explicados por mutaciones en el gen PTPN114 y hasta hace pocos meses el resto de los casos diagnosticados clínicamente solían quedar sin diagnóstico genético de certeza. Actualmente, una nueva mutación en el gen de la proteína SOS15 nos abre un campo más amplio al comprobarse su relación con el síndrome de Noonan en aproximadamente el 20 % de los casos que no presentan mutación del PTPN11.
El fenotipo asociado a los defectos de SOS1 tiene características distintivas como podemos comprobar en el caso que mostramos y que nos pueden facilitar la orientación de su diagnóstico genético.
OBSERVACIÓN CLÍNICAPaciente de 15 años remitido a los 2, 3/12 años de edad a nuestro servicio por presentar un fenotipo característico.
Entre sus antecedentes personales destacaba un embarazo a término, nacimiento por cesárea con un peso de recién nacido adecuado a su edad gestacional (2.800 g). Como incidencias presentó edema de pies y manos autolimitado en el período neonatal y precisó ingreso durante 9 días por un síndrome de adaptación pulmonar e ictericia neonatal con buena evolución. Refería haber estado en seguimiento por el servicio de cardiología por una estenosis pulmonar leve sin requerir tratamiento y que en la actualidad no se objetivaba. Presentaba un desarrollo psicomotor normal.
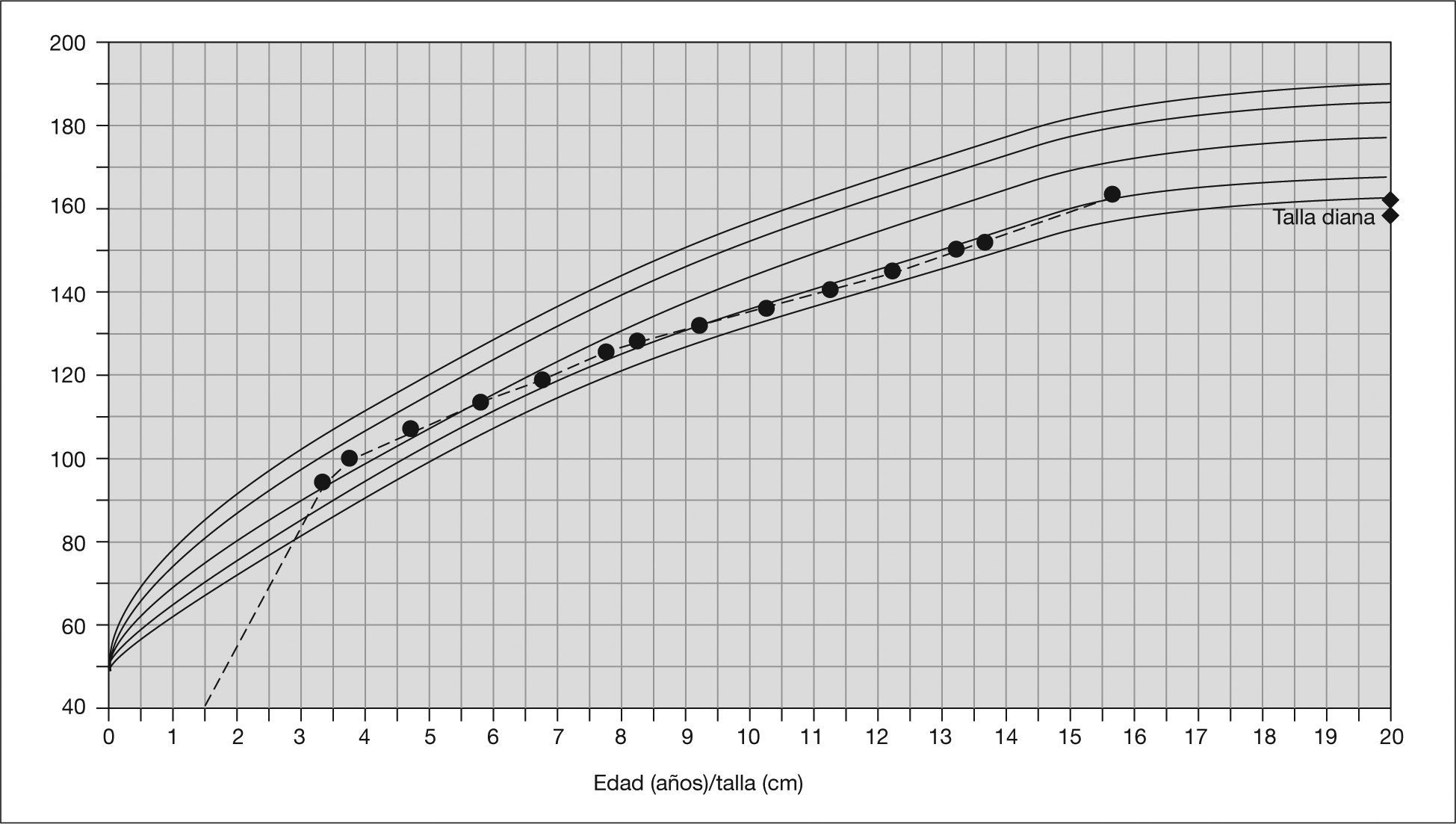
Entre los antecedentes familiares era llamativo el fenotipo materno con talla baja (147 cm), rasgos faciales similares a los del paciente, pectus excavatum y estenosis pulmonar leve. La talla diana del paciente era de 163 cm.
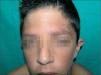
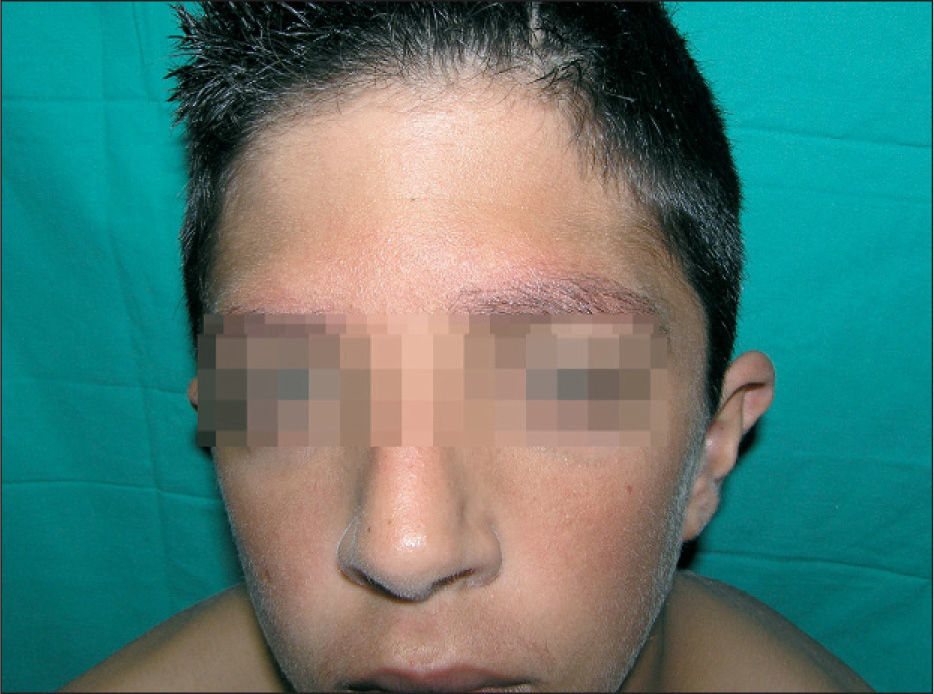
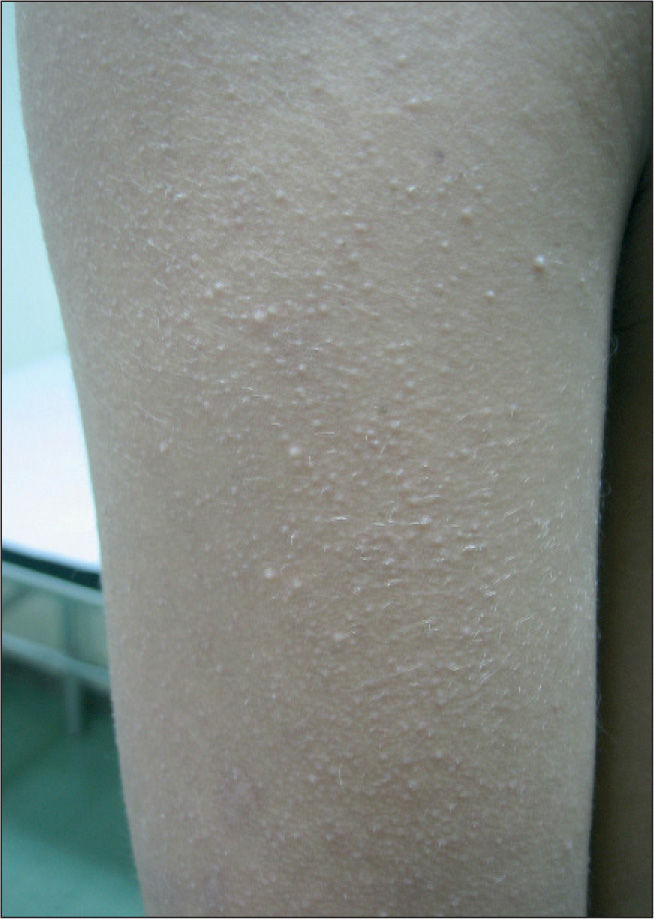
En la exploración física se objetivaban rasgos faciales característicos con pelo rizado, pabellones auriculares de implantación baja en anteversión, hendidura palpebral antimongoloide, hipertelorismo, epicanto, puente nasal plano y paladar ojival, cejas poco pobladas y queratosis pilar en cara acompañada de eritema malar y periocular, lo que conforma un ulerythema ophryogenes (fig. 1). También presentaba implantación baja de cabello, cuello corto, pterigium colli, tórax ancho, mamilas separadas y pies planos bilaterales. En la piel destacaba una importante queratosis pilar en brazos, piernas y tronco (fig. 2). Se observaba una hernia inguinal derecha pendiente de intervención con testes en escroto.
Con la sospecha clínica de síndrome de Noonan se solicitó cariotipo con resultado 46XY y estudio genético para dicho síndrome, que resultó negativo para las mutaciones del gen PTPN11, pero positivo para el gen SOS1, situado en el brazo corto del cromosoma 2; se encontró la mutación G(1297)A, que produce un cambio de aminoácido (Glu433Lys) en la secuencia del gen de la proteína SOS1, que resultará en un incremento de la actividad de la misma y de toda la cascada metabólica posterior.
Una vez conocido su diagnostico se procedió al seguimiento del paciente, que hasta la actualidad ha alcanzado una talla de 164 cm, con velocidad de crecimiento normal y un desarrollo puberal completo. Su desarrollo psicomotor ha estado dentro de límites normales durante todo el seguimiento.
DISCUSIÓNEl síndrome de Noonan es una entidad genéticamente heterogénea y en el 50 % de los casos es posible encontrar mutaciones en el gen PTPN116,7, que codifica una proteína denominada SHP-28. Esta proteína forma parte de diferentes vías que controlan el desarrollo proteico, y forma parte de estas mismas vías metabólicas la proteína SOS1, una de las dos proteínas SOS humanas, que codifica un factor RAS específico del intercambio del nucleótido guanina, imprescindible para la activación del receptor tirosinquinasa. Sabemos que la proteína SOS1 se encuentra en su estado basal autoinhibida debido a un complejo sistema de regulación intramolecular e intermolecular, cuando existe una mutación de su gen, como ocurre en nuestro caso, situado en el brazo corto del cromosoma 2 (locus 2p22-p21) desaparece la autoinhibición dando lugar a un incremento de la activación RAS que producirá un aumento de toda la cascada metabólica posterior5.
Desde el punto de vista fenotípico, se ha comprobado al agrupar pacientes con esta mutación, como ocurre en el estudio realizado por Tartaglia et al5, que aquellos con alteraciones del gen SOS1 tienen algunas características fenotípicas que los diferencian de otros síndromes de Noonan causados por mutaciones en el gen PTPN11 (tabla 1). Estos pacientes comparten con el resto su asociación con la estenosis pulmonar valvular o las alteraciones torácicas, cuello corto, pterigium y rasgos faciales típicos. Sin embargo, existen diferencias significativas en cuanto a la presencia de alteraciones ectodérmicas como la queratosis pilar, ulerythema ophryogenes o pelo rizado, mucho más frecuentes en casos con mutación del SOS1 y presentes todos ellos en nuestro paciente. Estas alteraciones cutáneas, aunque ya descritas en el síndrome de Noonan con anterioridad9, son frecuentes también en otros síndromes como el cardio-facio-cutáneo, con el que puede a menudo solaparse10. Es importante destacar cómo la afectación del crecimiento y el desarrollo psicomotor en estos niños no es tan acentuada; así comprobamos cómo en nuestro caso el paciente presenta un desarrollo psicomotor normal sin necesidad de educación especial, pubertad completa y una talla por encima de su talla diana (fig. 3). El hecho de presentar una talla normal y un desarrollo psicomotor adecuado también está asociado a este grupo de pacientes según los estudios realizados por Tartaglia et al5.
Estos casos de síndrome de Noonan asociados a mutaciones del gen SOS1 nos abren un nuevo campo diagnóstico y su variedad fenotípica nos ayuda a poder realizar un adecuado diagnóstico clínico y genético. Resaltamos el hecho de que la talla normal y el desarrollo psicomotor adecuado no deben ser criterios de exclusión del síndrome de Noonan, sino que nos pueden servir de guía para orientar adecuadamente el estudio genético como se comprueba en nuestro paciente. La presencia de una cardiopatía congénita, especialmente la estenosis pulmonar, junto con un fenotipo peculiar, debería orientar al diagnóstico de síndrome de Noonan y a la búsqueda de una etiología genética específica. De cualquier modo, aún son necesarios nuevos estudios para determinar otros genes causantes y conocer sus implicaciones en el diagnóstico diferencial y su pronóstico.
