



La trisomía parcial 4q es una enfermedad cromosómica rara, causada por una duplicación de una porción del cromosoma 4. En la mayoría de los casos resulta de una translocación balanceada de uno de los progenitores; siendo menos frecuente la aparición de novo. Presentamos un paciente con una duplicación parcial de novo del segmento distal del brazo largo del cromosoma 4 (q31, q35) asociada a translocación robertsoniana entre los cromosomas 14 y 21; asociación previamente no descrita. En la duplicación 4q no queda bien definida la relación entre el fenotipo y las partes del segmento duplicadas, aunque parece claro que anomalías renales y/o de pulgares son una manifestación característica. Revisamos la literatura médica, y de los casos previamente descritos con trisomía q31-35 concluimos que esta región del cromosoma 4 también estaría comprometida en constituir el “síndrome de la trisomía parcial 4q” o “síndrome aurículo-acro-renal”
The partial trisomy 4q is a strange chromosomal illness. This illness is caused by the duplication of a portion of chromosome 4. In most of the cases, it is the result of a balanced translocation in one of the progenitors. The “de novo” appearance is less common. We present a patient with a partial “de novo” duplication in the distal segment of the long arm of chromosome 4 (q31, q35), in association with Robertsonian translocation between chromosomes 14 and 21. This association has not been described previously. In the 4q duplication, the relationship between the phenotype and the parts of the duplicated segment is not well defined, although it seems clear that the renal anomalies and/or thumbs abnormalities are a characteristic manifestation. We have reviewed the literature and, of the cases previously described with trisomy q31-35, we came to the conclusion that this region of chromosome 4 may also be involved in constituting the “Syndrome of partial trisomy 4q” or Auriculo-acro-renal Syndrome”.
La duplicación 4q es un trastorno cromosómico poco frecuente del que se han descrito 60–70 casos en la literatura médica. En el 90 % se debe a una translocación equilibrada de uno de los padres, y es más rara la aparición de novo. La región duplicada es muy variable en los distintos casos publicados, pero parecen identificarse unos rasgos fenotípicos peculiares que asociarían de forma casi constante malformaciones del pulgar y malformaciones renales.
Presentamos un paciente con una duplicación parcial de novo del segmento distal del brazo largo del cromosoma 4 (q31, q35) asociado a translocación robertsoniana entre los cromosomas 14 y 21, hasta ahora no referida en la literatura médica. Sólo hemos encontrado a otros dos hermanos referidos por Otsuka1 con una duplicación de cromosoma 4 que afecta a las bandas q31 y q35 y similar fenotipo. Discutimos el caso y revisamos la literatura médica.
OBSERVACIÓN CLÍNICASegundo hijo de padres sanos, jóvenes, no consanguíneos. Parto a término, Apgar 9/10; peso: 2.540g (P10); longitud: 48cm (p25-50); perímetro cefálico 31 (< p10). Exploración: fenotipo peculiar con microcefalia, pabellones auriculares displásicos grandes, de implantación baja con antehélix prominente y plano, hélix picudo con hipoplasia de trago; hendiduras palpebrales antimongoloides; filtrum corto con ángulo de la boca hacia abajo; barbilla picuda. Desproporción, con tronco más largo que las extremidades. Auscultación cardiopulmonar soplo sistólico I/VI. Abdomen normal. Neurológicamente la fuerza y motricidad son normales, discreta hipotonía axial sin hipertonía de extremidades, audición y visión normales. Aparato locomotor: implantación baja de pulgares aunque sobrepasan el pliegue palmar, sin hipoplasia de eminencia tenar y clinodactilia. Genitales: criptorquidia derecha.
Exploraciones complementarias- –
Cariotipo: translocación robertsoniana entre los cromosomas 14 y 21, así como una duplicación directa del cromosoma 4 entre las bandas q31 y q35. Cariotipo 45,XY, der (14;21) (q10;q10), dup (4) (q31 q35).
- –
Cariotipo de los padres: normales.
- –
Ecocardiograma: normal.
- –
Radiografía de ambas manos: clinodactilia del quinto dedo y pulgares bifalángicos bilaterales.
- –
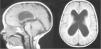
Resonanacia magnética cerebral: dilatación de ventrículos con ventriculomegalia no hipertensiva, importante disminución sustancia blanca periventricular, cuerpo calloso atrófico (fig. 1).
- –
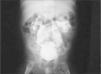
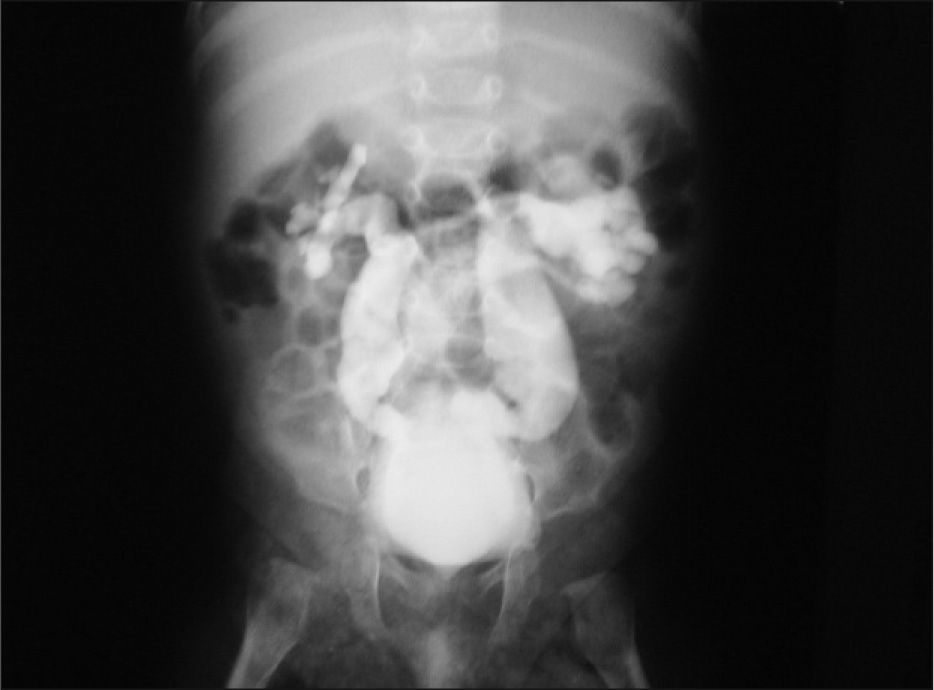
Estudios nefrológicos: ecografía renal al mes de vida que muestra ectasia ureteropielocalicial bilateral con ríñones de tamaño y ecoestructura normal. Cistouretrografía miccional seriada (CUMS) al mes de vida: ausencia de RVU (reflujo vésico ureteral). Múltiples divertículos vesicales. Uretra normal. Urografía intravenosa (UIV) a los dos meses de edad: ureterohidronefrosis bilateral (fig. 2). Renograma diurético MAG3 al 5¿ mes de vida: riñones morfológicamente normales. Curvas renográficas con ectasia no obstructiva bilateral y función relativa del 50 % para cada riñón. Urodinamia a los 9 meses: vejiga de escasa capacidad con contracciones no inhibidas del detrusor a partir de 25ml de infusión, que generan presiones intravesicales de 100cm H2O con continuos escapes.
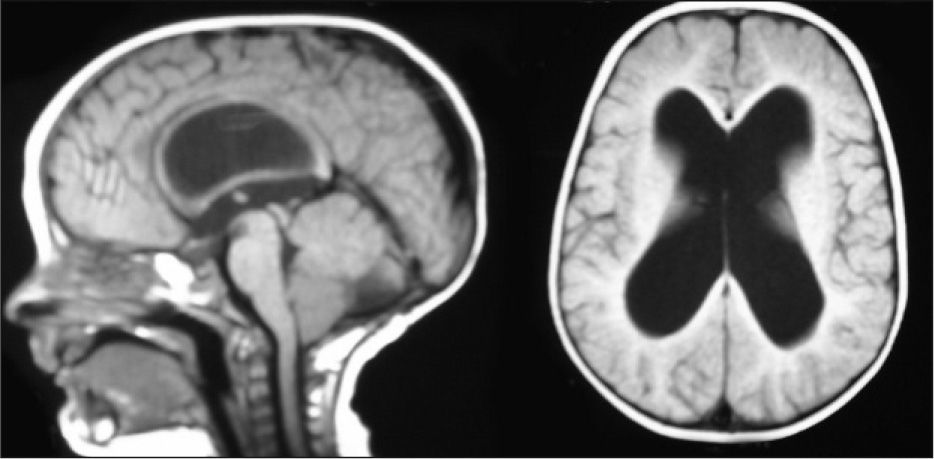
Retraso de hitos motores. A los 6 años el fenotipo es más acentuado y se perfila mejor la displasia auricular, las hendiduras palpebrales antimongoloides con hipertelorismo, raíz y puente nasal anchos y el ángulo de la comisura bucal hacia abajo (fig. 3), presenta lenguaje normal y alteración de motricidad fina.
Evolución urológica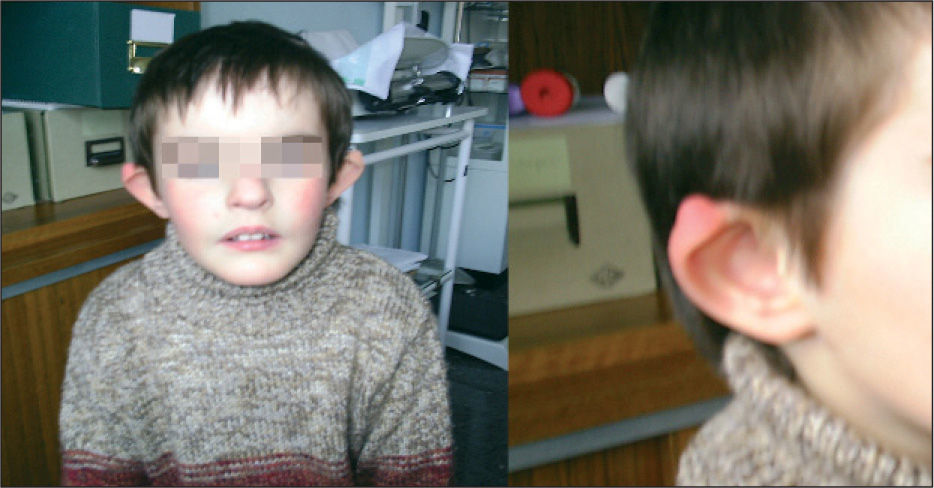
Megauréter bilateral no obstructivo secundario a vejiga neurógena no neurogénica de carácter hipertónico. Evolución favorable del riñón derecho, con tratamiento anticolinérgico con oxibutinina oral. En el riñón izquierdo se objetivó un empeoramiento de la función renal relativa, que disminuyó hasta el 40 %, con un patrón obstructivo en la curva excretora. Precisó ureteroneocistostomía tipo Cohen, posteriormente el crecimiento renal fue adecuado y recuperó la función del riñón izquierdo.
DISCUSIÓNPresentamos un paciente con duplicación parcial de novo del segmento distal del brazo largo del cromosoma 4 (q31, q35) asociado a translocación robertsoniana entre los cromosomas 14 y 21.
El cariotipo de una translocación robertsoniana tiene 45 cromosomas; sin embargo, se dice que es equilibrada (no hay pérdida ni ganancia de material genético), y no tiene efectos fenotípicos. La más frecuente es la translocación 14;21 que fue una de las alteraciones genéticas que presentó nuestro paciente y que originó un fórmula con 45 cromosomas: 45,XY, der (14;21) (q10;q10). En las anomalías cromosómicas estructurales se puede producir una pérdida o ganancia de material genético (delección o duplicación) de uno o varios segmentos cromosómicos. El cambio será desequilibrado y, por tanto, se traducirá en alteraciones fenotípicas. En nuestro caso encontramos una duplicación directa del cromosoma 4 entre las bandas q31 y q35, que originó la alteración fenotípica y las malformaciones cerebrales, renales y óseas.
En la trisomía parcial 4q se han descrito otras alteraciones genéticas asociadas en más del 10% de los casos: monosomía 5p2, monosomía 9p3, monosomía parcial 18q4, monosomía 21q5 trisomía 9p6, trisomía parcial 137, etc., ya que el material cromosómico extra del 4q puede translocarse desde otro cromosoma e inducir en este último una monosomía, y con superposición clínica por ambas alteraciones dificultar la identificación y relación fenotipo-genotipo. Se ha reservado el término trisomía parcial “pura” 4q cuando no asocia ninguna otra alteración genética.
Hasta ahora no hemos encontrado referido en la literatura médica la asociación de esta translocación robertsoniana der (14; 21) (q10;q10) en la duplicación del cromosoma 4, como la que presentó nuestro paciente; pero, en cualquier caso, esta asociación no contribuyó a sus características fenotípicas y podemos considerarla a este efecto una trisomía “pura”. En la etiología de estas alteraciones cromosómicas con varias asociaciones genéticas se ha encontrado hasta en el 90 % una translocación equilibrada de uno de los padres, como consecuencia de una alteración en la segregación de diferentes translocaciones recíprocas, y es más rara la aparición de novo3,6,7; en otros casos se ha atribuido a alteración de la segregación durante la meiosis en la madre8, como sospechamos que ocurrió en nuestro caso.
La duplicación 4q es un trastorno cromosómico poco frecuente. La clínica es variable dependiendo del segmento duplicado. El peso al nacer es bajo en la mitad de los casos. Las alteraciones craneofaciales más frecuentes son: microcefalia y frente inclinada (75 %), hipertelorismo (75%), epicantus (40%), fisuras palpebrales antimongoloides (35%), puente nasal largo (40%), cuello corto 60%, ángulo de la boca hacia abajo (35%), implantación baja de los pabellones auriculares con antehélix prominente e hipoplasia de trago (90 %), barbilla puntiaguda y micrognatia. La mitad de los pacientes presentan cardiopatías congénitas. Alteraciones genitourinarias en el 30 % (riñón en herradura, hipoplasia renal, reflujo ureterovesical con o sin hidronefrosis). Hernia inguinal o umbilical hasta en el 30 % y en varones, criptorquidia9,10. Neurológicamente se hace referencia a la hipotonía o hipertonía (60 %) y epilepsia, pero se describen pocas malformaciones cerebrales en pruebas de neuroimagen.
Actualmente la relación entre el fenotipo y la parte del segmento duplicada del brazo largo del cromosoma 4 no queda bien establecida. Esta relación es más difícil en los casos en los que la trisomía es resultado de una translocación equilibrada familiar porque en ellos suele existir una monosomía concomitante. Sin embargo, algunos autores han sugerido que el fenotipo es muy sugerente de constituir un auténtico síndrome de trisomía 4q cuando se presenta “puro”. Las regiones cromosómicas comprometidas en esta trisomía son amplias, entre las bandas q21 y q3211. Para Lundin et al12 la región 4q27 y 4q31 sería la preferentemente comprometida en constituir el síndrome de la trisomía 4q. Sin embargo, otros autores, como Zollino et al13 identifican la región 4q22-23 también comprometida en el síndrome de trisomía parcial 4q y Battaglia et al14 incluye la región 4q25-28. La relación entre la trisomía 4q asociada a la patología renal y las malformaciones del pulgar han sido discutidas, y actualmente se consideran manifestaciones características de la duplicación del cromosoma 4, para cuya denominación se ha propuesto el término “acro-renal”. Para otros autores, se necesitan más investigaciones que delimiten el espectro clínico de las características esenciales para definir el síndrome de la trisomía parcial 4q15,16.
Aunque para Lundin et al12 la región 4q27 y 4q31 sería la comprometida en constituir el síndrome de la trisomía 4q, hemos identificado rasgos dismórficos típicos del llamado “síndrome de trisomía 4q” en nuestro paciente. La mayoría de estos rasgos se identifican también en los otros dos hermanos asiáticos con trisomía q31-35 referidos por Otsuka1, pese a las diferencias raciales, por lo que pensamos que esta región del cromosoma 4 también estaría comprometida en constituir el síndrome de la trisomía 4q.
En el análisis comparativo de los casos publicados que realiza Elghezal et al17 se sugiere que la región 4q31-q33 podría estar envuelta en el desarrollo de las características dismórficas típicas de esta duplicación, y la banda distal 4q35 podría afectar al desarrollo de microcefalia, retraso mental grave y retraso de crecimiento. Tanto nuestro paciente, como en los otros dos pacientes referidos con afectación citogenética de la banda distal 4q31-q35, presentaron un retraso psicomotor leve o moderado. Estas observaciones no apoyan que la banda q35 sea exclusiva en el desarrollo de retraso mental18.
En conclusión, a la vista de nuestro caso y los descritos anteriormente, las discusiones previas entre la relación fenotipo/genotipo parecen aclararse si se excluyen los casos que cursan con monosomía asociada, ya que en las formas puras de trisomía parcial 4q al menos entre las bandas q22-q35, sí parece identificarse un síndrome específico. El espectro clínico de las características esenciales para definir el síndrome de la trisomía parcial 4q consistirían en retraso mental de leve a grave; fenotipo peculiar, en el que destacan los pabellones auriculares displásicos con antehélix grande, hendiduras palpebrales antimongoloides y desviación hacia debajo de la comisura bucal, malformaciones de los dedos de las manos, predominantemente el pulgar y de forma casi constante malformaciones renales y/o urológicas con criptorquidia en varones. Proponemos así la denominación “aurículo-acrorenal” para el síndrome de la trisomía parcial 4q, ya que las malformaciones del pabellón auricular son también típicas y constantes.
