
Sr. Editor:
El síndrome de Noonan aparece en uno de cada 1.000-2.500 recién nacidos, tiene un carácter autosómico dominante y se caracteriza por presentar talla baja, dismorfias faciales, defectos cardíacos y criptorquidia en varones. El gen responsable (PTPN11) ha sido recientemente identificado, y explica entre el 30 y el 50 % de los casos diagnosticados por la clínica 1.
Paciente de 5 años y 2 meses que consulta en nuestro servicio desde los 3 años por presentar un fenotipo característico. No antecedentes familiares de interés. Cesárea por oligoamnios, peso de recién nacido 3.350 g. Desarrollo psicomotor normal, estenosis válvula pulmonar intervenida a los 7 meses. Orquidopexia bilateral a los 2 años y 7 meses. Laringotraqueomalacia. Púrpura trombocitopénica a los 2 años y 9 meses. Talla diana 172,2 cm. En la exploración física inicial destacaba una talla de 87,9 cm (2 sds), junto a pabellones auriculares de implantación baja y rotados, hendidura palpebral antimongoloide con disminución de la apertura, hipertelorismo, epicanto, puente nasal plano, pterigium colli, tórax en quilla y discreta dificultad bilateral en la extensión del codo. Testes en escroto. Cicatrices de orquidopexia y esternotomía. Aporta cariotipo 46XY. Actualmente presenta una talla de 103,1 cm (1,86 sds).
Se realiza estudio genético encontrándose la mutación A(923)G, que produce un cambio de aminoácido (Asn308Ser) en la secuencia del gen PTPN11 en el cromosoma 12.
El síndrome de Noonan fue descrito por primera vez en 1963 por Noonan y Ehmke 2 al presentar 9 casos de niños con estenosis valvular pulmonar que asociaban otras malformaciones sugestivas de síndrome de Turner, aunque la primera descripción fenotípica concordante data de 1883 por Kobilinsky.
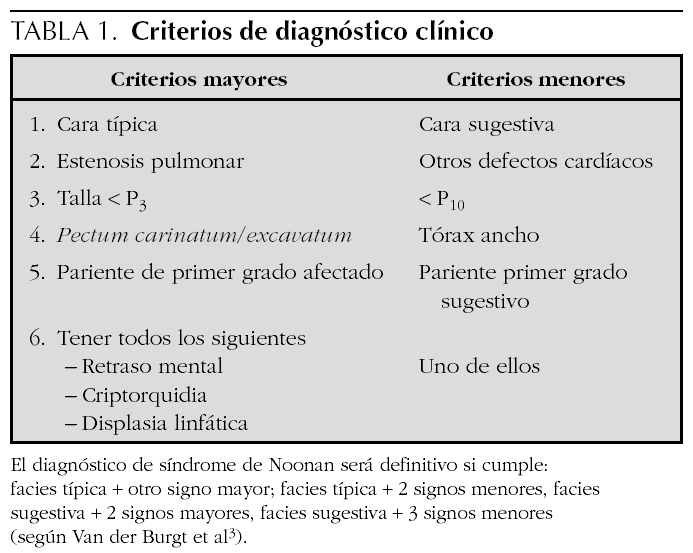
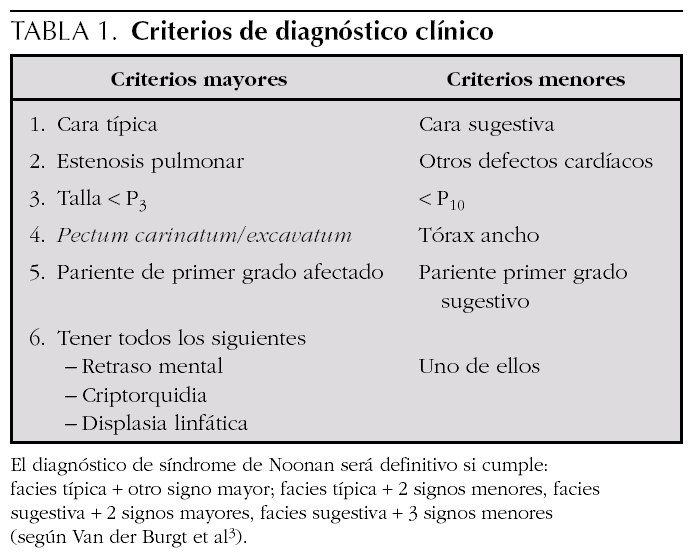
El síndrome de Noonan tiene una herencia autosómica dominante y se caracteriza por talla baja proporcionada, dismorfias faciales, como hipertelorismo, fisuras palpebrales antimongoloides, orejas de implantación baja y rotadas, implantación baja del pelo, cubitus valgus, defectos cardíacos congénitos (especialmente estenosis valvular pulmonar y miocardiopatía hipertrófica), deformidades torácicas y vertebrales, retraso mental leve alteraciones hematológicas, retraso puberal y criptorquidia en varones. Hay unos criterios clínicos propuestos por Van der Burgt et al 3 que facilitan el diagnóstico (tabla 1). El síndrome de Noonan es genéticamente heterogéneo con mutaciones en el gen PTPN11 en el 50 % de los casos 4,5. El gen PTPN11 está localizado en el cromosoma 12q24.1 y codifica el no receptor protein-tirosín-fosfatasa SHP-2. Se han descrito mutaciones en los exones 1, 2, 3, 4, 7, 8, 12, 13 y 14, siendo el 80 % en el 3 y 8. La SHP-2 forma parte de diferentes vías que controlan el desarrollo proteico, especialmente la valvulogénesis de la válvulas semilunares cardíacas. Estas mutaciones llevan a un aumento de actividad de este gen. La prevalencia de estas mutaciones es mayor en los casos familiares que en los esporádicos. Este gen también se ha relacionado con otras enfermedades como la asociación Noonan/neurofibromatosis tipo 1, síndrome de LEOPARD 6, síndrome cardio-facio-cutáneo y diferentes tipos de leucemias 7,8 y tumores sólidos 9. Se ha demostrado una asociación significativa entre la estenosis valvular pulmonar y la mutación en el gen PTPN11 en pacientes con síndrome de Noonan, mientras que la miocardiopatía hipertrófica es más prevalente entre los que no tienen mutaciones en ese gen. Para el resto de alteraciones clínicas, no se han demostrado diferencias significativas 10,11.
El diagnóstico del síndrome de Noonan se realiza mayoritariamente por la clínica, pero es posible confirmar el diagnóstico genéticamente hasta en el 50 % de los casos. Son necesarios nuevos estudios para determinar otros genes causantes de este síndrome y saber las posibles implicaciones de estos en el diagnóstico diferencial y pronósticas.
Correspondencia: Dr. J.M. Lloreda-García.
Servicio de Endocrinología Pediátrica.
Hospital Materno-Infantil Carlos Haya.
Arroyo de los Ángeles, s/n. 29011 Málaga. España.
Correo electrónico: jmlloreda@yahoo.es
