

Sr. Editor:
La histiocitosis de células de Langerhans (HCL) es una enfermedad muy infrecuente, cuya forma congénita aún lo es menos. Esta última, descrita inicialmente como enfermedad de Hashimoto-Pritzker, involuciona en gran parte de los casos de forma espontánea 1; dado que esto no siempre es así, es necesario un seguimiento estricto del paciente para descartar enfermedad multisistémica y decidir sobre el tratamiento a aplicar. Esto ocurre en el caso que presentamos, de una histiocitosis congénita, que, finalmente, requirió tratamiento en la Unidad de Oncología Pediátrica.
Presentamos una recién nacida a término y sin incidencias perinatales, que en el momento del nacimiento presenta un exantema maculovesicular (ampolloso en algunas zonas), generalizado, que afecta a palmas y plantas, predominante en cara y tronco. Inicialmente las lesiones sugerían una varicela o herpes congénito, una forma de impétigo o, en menor medida, una melanosis pustulosa neonatal, sin confirmarse ninguna de ellas.
Al mes de vida el exantema evoluciona a papulodescamativo y petequial, asociando estancamiento ponderal (pérdida de 180 g de peso), rechazo de las tomas, irritabilidad y hepatomegalia de 3 cm b.r.c. Asocia anemia, con Hb 8,5 g/dl (9,5-14 g/dl) y VCM 100,8 fl (85-123 fl), con discreta leucocitosis (17.270), con fórmula anodina y sin atipias morfológicas, sin alteración en la serie plaquetaria y VSG 73 mm/h. Resto de parámetros bioquímicos normales.
La biopsia cutánea confirma la sospecha diagnóstica: HCL, con infiltrado dérmico difuso en dermis papilar constituido por células de hábito histiocitario con citoplasma eosinófilo y núcleo arriñonado. Los citoplasmas son PAS positivos y el estudio inmunohistoquímico demuestra positividad de las células para el antígeno de superficie CD1 y para la proteína S-100.
Con este diagnóstico se continúa el estudio para valorar la afectación sistémica de la enfermedad, incluyendo ecografía abdominal, radiografía de tórax, serie ósea y analítica general (con función hepática y renal), sin apreciarse alteraciones, por lo que, sospechando una enfermedad de Hashimoto-Pritzker (entidad normalmente autoinvolutiva), se adopta, inicialmente, una actitud expectante. Pero ante la persistencia del estancamiento ponderal y de la anemia (8,2 g/dl), con aparición de febrícula al mes y medio de vida, se decide ampliar estudio en la Unidad de Oncología de referencia.
Ya en dicha unidad, con 2 meses de vida, el estudio realizado (ecografía abdominal, TC de cráneo, de región mastoidea y de tórax, así como biopsia de médula ósea) sigue sin evidenciar afectación sistémica hasta que al repetir una segunda serie ósea se descubre una lesión osteolítica en radio (fig. 1), la cual junto con la aparición de hipoproteinemia (2 g/dl), hizo considerar el grado de afectación sistémica tributario de iniciar tratamiento con quimioterapia (prednisona, vinblastina y metotrexato). Con ésta, la evolución clínica inicial es favorable, desapareciendo las lesiones cutáneas, cediendo la irritabilidad, tolerando la alimentación y ganando peso. Transcurridos casi 4 meses con tratamiento, fallece por afectación intercurrente aguda bronconeumónica no filiada (la familia no concedió la necropsia).
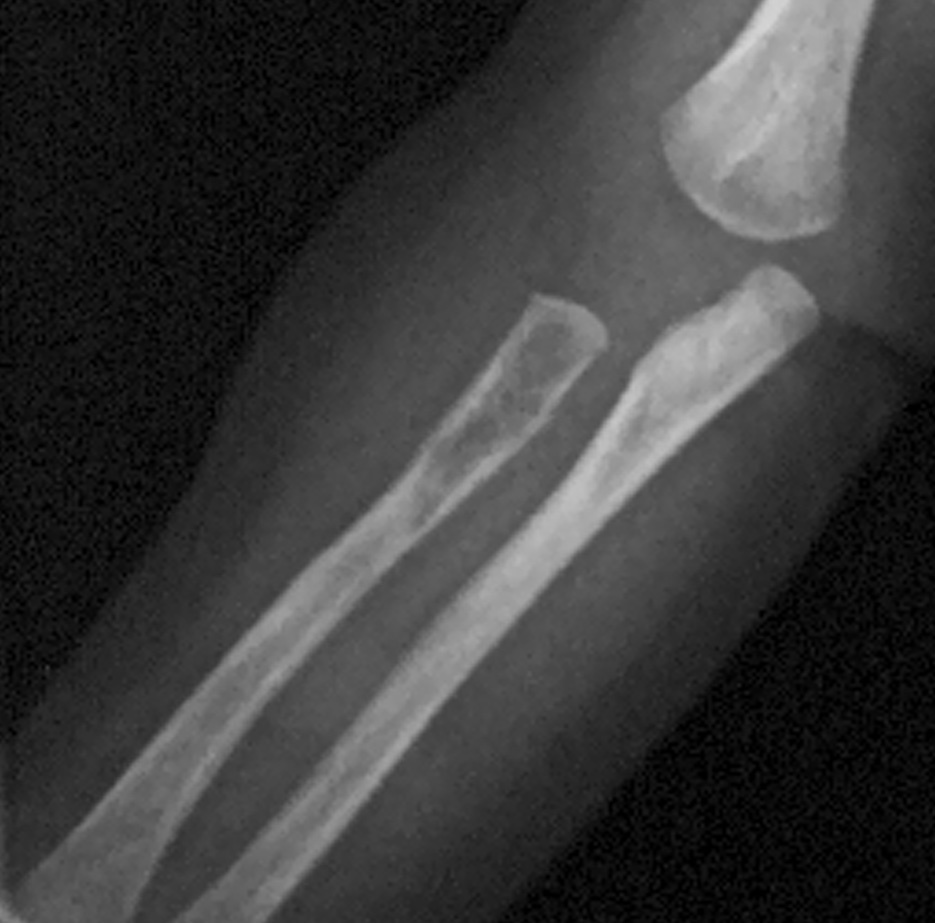
Figura 1. Lesión osteolítica en radio.
La forma congénita de la HCL es muy poco frecuente y polimorfa. En 1973 Hashimoto y Pritzker 2 la describieron como una entidad autorresolutiva y de afectación sólo cutánea. Posteriormente se han descrito formas congénitas más heterogéneas que complican el diagnóstico diferencial en esa primera fase de la enfermedad.
Se puede presentar ya en el momento del nacimiento o en el período neonatal. El órgano con mayor frecuencia afectado es la piel (hasta en un 82 %) 1, las lesiones suelen ser múltiples, como pápulas o nódulos 3, o en forma de erupción papulovesicular generalizada 4 que evolucionan hacia la descamación o al exantema petequial como en nuestro caso, pero también se han descrito lesiones solitarias papulonodulares 5.
El diagnóstico diferencial debe realizarse con otras erupciones del recién nacido como la varicela, el herpes simple, la melanosis pustulosa neonatales y la candidiasis congénita. Se llegará al diagnóstico de HCL a través de la biopsia cutánea, donde se observarán gránulos de Birbeck a través de la microscopia electrónica, y en las tinciones de inmunohistoquímica se confirmará la presencia de proteína S-100 y el Ag CD1 será positivo.
Los casos congénitos descritos en la bibliografía, en su mayoría, son formas exclusivamente cutáneas y autolimitadas (enfermedad de Hashimoto-Pritzker). Aunque también se han descrito formas que han asociado participación de otros órganos, como el hueso 6 (en forma de granuloma eosinófilo), el pulmón 7, el hígado (con elevación de GOT-GPT), la médula ósea 8,9 (con leucopenia, trombocitopenia, infiltración monocitaria atípica) y que aún así finalmente, regresan de forma espontánea. Así mismo, se describen pacientes que desarrollan rápidamente afectación multisistémica. En un estudio retrospectivo de 19 pacientes con HCL diagnosticados en el período neonatal 1, sólo en un tercio la enfermedad estuvo limitada a la piel; sin encontrar diferencias en las características morfológicas de las lesiones mucocutáneas entre éstos y aquéllos con afectación multisistémica, pero es de resaltar que en todos los que tuvieron un curso autolimitado las lesiones cutáneas estaban presentes ya al nacimiento. Cuando apareció afectación multisistémica la mortalidad llegó hasta el 16 %.
El desafío, pues, surge al intentar determinar hasta cuándo esperar la involución espontánea, típica de esta forma de presentación, para evitar así iatrogenia innecesaria, lo que supone saber ponderar el grado de afectación sistémica que indicaría una progresión definitiva.
Las formas congénitas de HCL, cuya afectación al inicio sea exclusivamente cutánea, en sus primeras descripciones parecían garantizar un buen pronóstico, con involución espontánea. Nuestro paciente, sin embargo, ejemplifica la necesidad de un seguimiento a medio-largo plazo en estos casos, visto su potencial de diseminación en un lapso de tiempo que aún es incierto 10.
Correspondencia: Dr. M.A. Fuentes Castelló.
Camí de l'Almazara, 11. 03203 Elche. España.
Correo electrónico: mafcros@yahoo.es
