


Sr. Editor:
El hamartoma fibroso de la infancia (HFI), un tumor raro y único en los niños fue descrito por Reye en 1956 1.
Presentamos el caso de un paciente varón de 6 meses fue remitido a nuestra consulta para valoración de un tumor congénito, asintomático, en la espalda. El tumor permanecía estable desde el nacimiento y la piel que lo recubría había ido desarrollando hipertricosis. En la exploración física en el centro de la espalda presentaba bajo una piel de características normales, excepto por el aumento del vello, un nódulo subcutáneo, firme, no adherido a planos profundos de 4 × 3,7 cm (fig. 1). Bajo anestesia local la lesión fue extirpada. El estudio histopatológico reveló la presencia de una proliferación mal delimitada en el tejido celular subcutáneo compuesto por miofibroblastos, pequeños nidos de células mesenquimales dispersas y grasa madura entremezclada (fig. 2). Dos años después el paciente permanece asintomático.
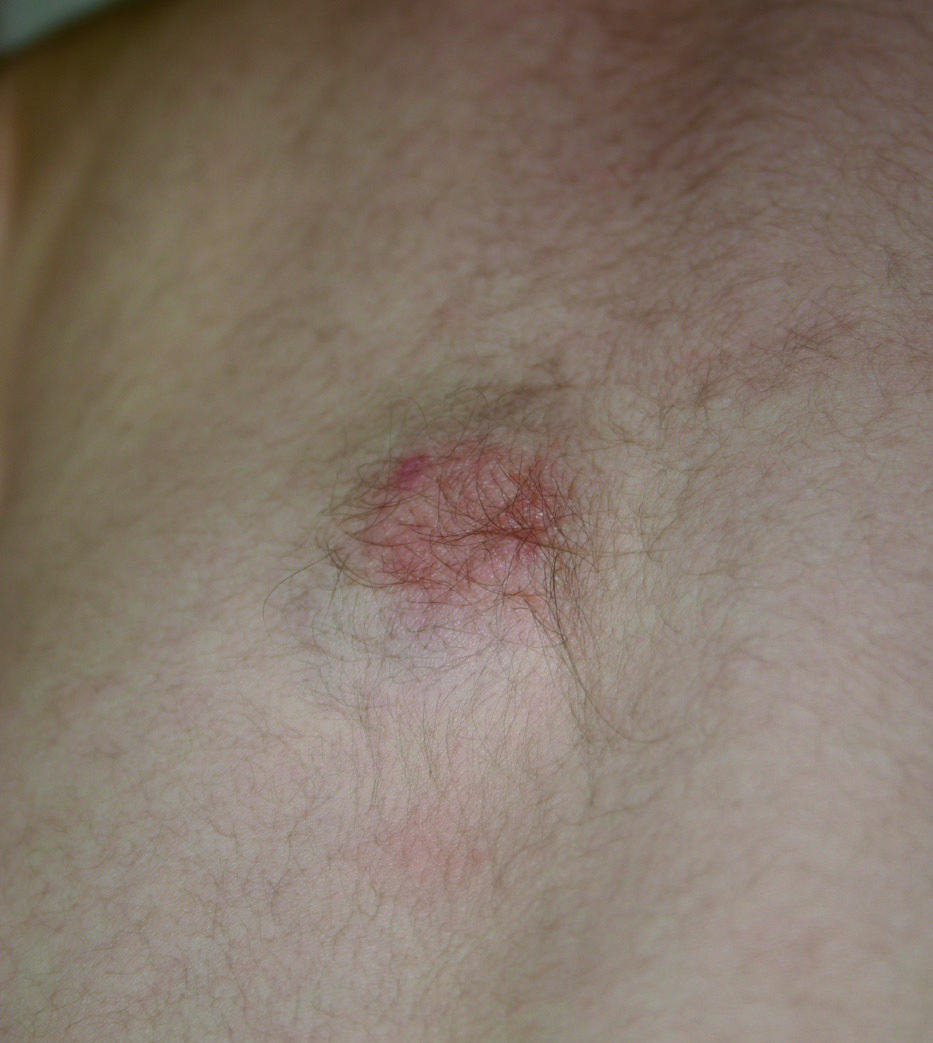
Figura 1. En el centro de la espalda nódulo duro, firme al tacto, no adherido a planos profundos.
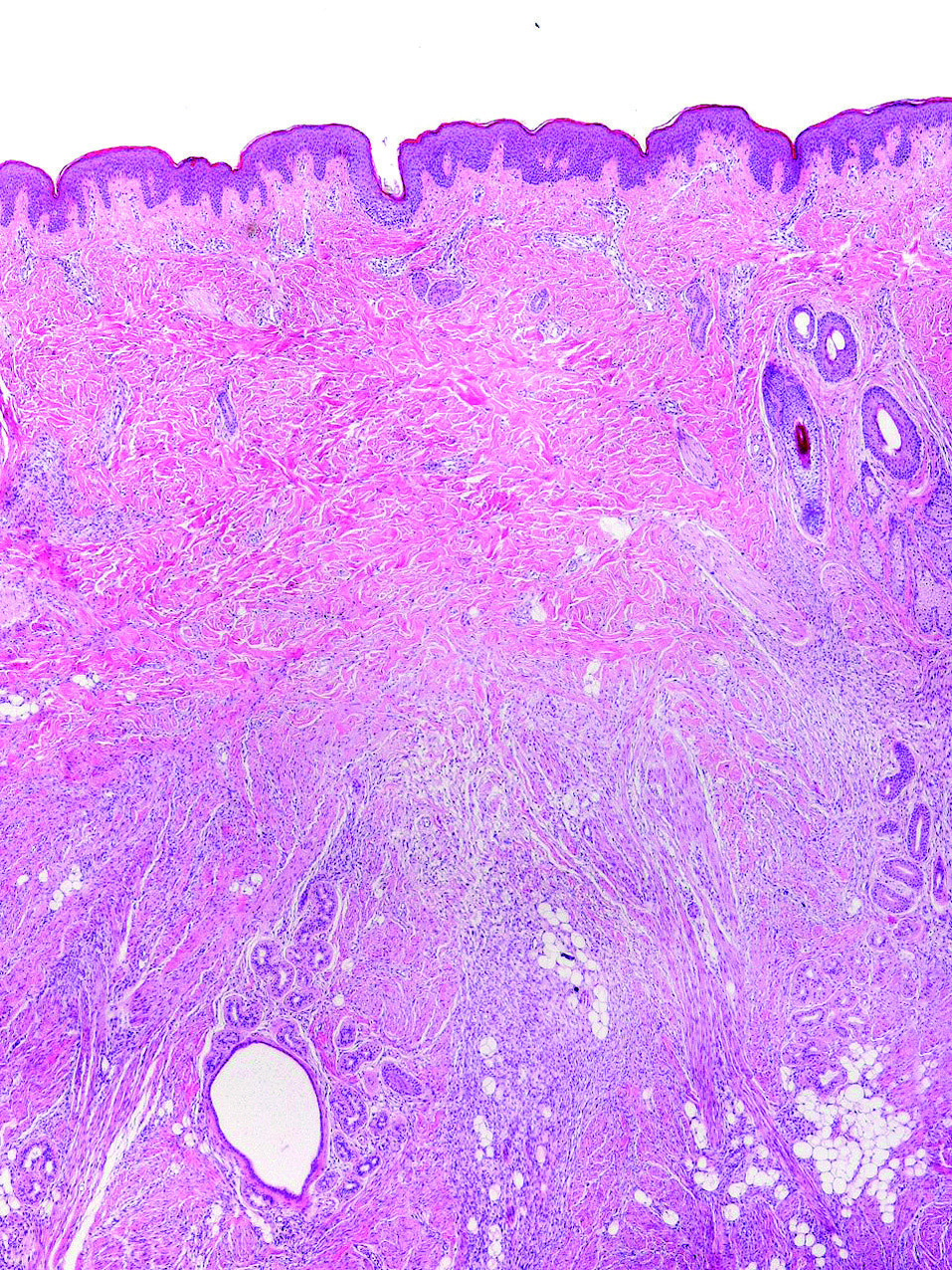
Figura 2. Proliferación compuesta por miofibroblastos, células mesenquimales y grasa de manera dispersa.
El HFI es un tumor raro, benigno, persistente que aparece durante los primeros 2 años de vida como un nódulo subcutáneo firme, indoloro, de crecimiento lento que afecta predominantemente a los varones. Sólo unos pocos casos presentan cambios en la piel que los recubre como alteraciones en la pigmentación, hiperplasia de glándulas ecrinas e incremento del pelo 2,3. El tumor se localiza habitualmente en el tronco, especialmente en la axila y extremidades superiores, y de manera más infrecuente en extremidades distales, cabeza, cuello y escroto 4.
Histológicamente se trata de un hamartoma, que por definición consiste en la proliferación desorganizada de tejidos normales en la localización en que se producen.
El tumor consta de 4 componentes principales en diferentes proporciones: a) fascículos de miofibroblastos, de color eosinófilo pálido y actin positivo, con núcleos estrechos y ondulados; b) pequeños cúmulos de células fusiformes indiferenciadas en estroma mixoide; c) tejido colágeno desordenado con vasos y células inflamatorias, y d) islas de adipocitos maduros 5.
El diagnóstico diferencial más común incluye la fibromatosis juvenil, el hamartoma de músculo liso, lipomas, sarcomas, dermatofibromas y neurofibromas 6.
El tratamiento de elección es la extirpación local, con poca tendencia a la recurrencia incluso en exéresis con sección tumoral.
Nosotros presentamos un caso de este raro tumor sobre el que existen pocas publicaciones en la que se muestre macroscópicamente la lesión.
Correspondencia: Dr. J. Santos-Juanes.
Servicio de Dermatología II.
Hospital Central de Asturias. Oviedo. España.
Correo electrónico: jsantosj@hca.es.
