


Sr. Editor:
El síndrome de Kallmann-Maestre de San Juan- Morsier (SKMSJ)1 es la combinación de hipogonadismo hipogonadotrópico y anosmia o hiposmia. Se debe a una deficiencia en la liberación de la hormona hipotalámica Gn-RH (gonadotropin-releasing hormone 'hormona liberadora de gonadotropinas'), probablemente por un fallo en la migración embrionaria de las neuronas productoras de Gn-RH, junto con ausencia o hipoplasia de los nervios y bulbos olfatorios. Puede asociar otras malformaciones.
Caso 1: varón de 16 años que consulta por hipogenitalismo. Los antecedentes familiares son desconocidos. Entre los antecedentes personales, destaca que a los 14 años se le diagnosticó e intervino de una hernia diafragmática izquierda que contenía la cámara gástrica, el bazo y el colon descendente en la cavidad torácica. Refería anosmia desde la infancia. En la exploración se observa peso de 44,2kg (percentil [P]3), talla de 155cm (P10). Los genitales son masculinos con micropene (pene de 2 × 1cm) y criptorquidia bilateral (testes inguinales de 1 cc, que se pueden descender al escroto). Presenta estadio i de Tanner. En las pruebas complementarias se observa cariotipo 46XY; testosterona total (T) de 0,1ng/ml (valor normal [VN]: 2,7¿8,3); test de Gn-RH (100¿g intravenosa [i.v.]: 0¿, 20¿, 60¿); FSH (follicle-stimulating hormone 'hormona foliculoestimulante o folitropina') inferior a 1,0; 2,1; 3,7mU/ml; LH (luteinizing hormone 'hormona luteinizante o lutoestimulante') inferior a 1,0; 2,7; 3,4mU/ml. El resto de la función adenohipofisaria se presenta sin alteraciones. La edad ósea es de 12 años (según el atlas de Greulich y Pyle). La resonancia magnética (RM) muestra aplasia de bulbos olfatorios (figura 1). Se realiza orquidopexia bilateral y se inicia tratamiento con gonadotropina coriónica humana (HCG) (1.000 U semanales) que el paciente rechaza a la tercera dosis, por lo que se sustituye por enantato de testosterona, inicialmente a una dosis de 50mg cada 3 meses y progresivamente se aumentó hasta llegar al mantenimiento con 100mg mensuales. Actualmente tiene un estadio IV de Tanner, con pene de tamaño normal y testes de 4 cc.
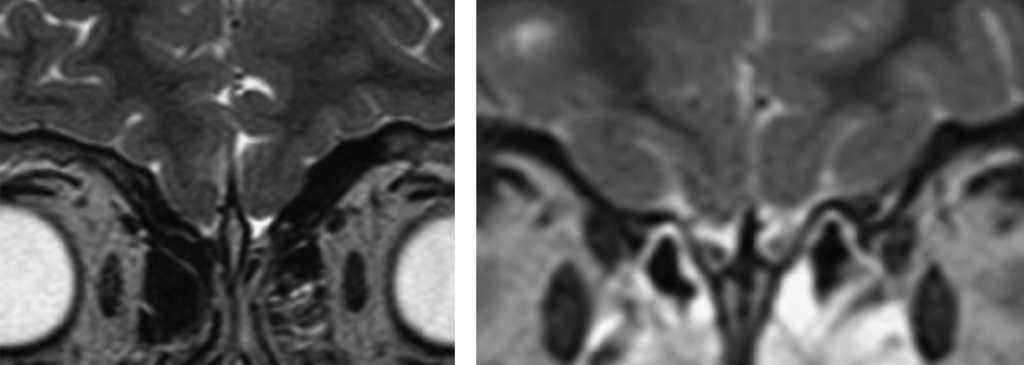
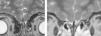
Figura 1. A la izquierda, resonancia magnética (RM) con ausencia de bulbos olfatorios; a la derecha, RM normal.
Caso 2: varón de 7 años que consulta por micropene. Entre los antecedentes personales, destacan sordera bilateral, anosmia, comunicación interauricular, valvulopatía aórtica y tricuspídea. Los antecedentes familiares no están relacionados. En la exploración se observa peso de 15,8kg (P50), talla de 97,5cm (P3). Presenta micropene (pene de 1,5 × 1cm) y criptorquidia bilateral (testes inguinales de 1cc). Tiene estadio i de Tanner. En las pruebas complementarias se observa FSH inferior a 1,0mU/ml; LH inferior a 1,0mU/ml; T inferior a 0,1ng/ml (VN: 2,7 a 8,3). El resto de la función adenohipofisaria resulta normal. La edad ósea es de 5 años (Greulich y Pyle). La ecografía abdominopélvica muestra testes inguinales, con diámetros de 12mm (derecho) y 11mm (izquierdo). Su cariotipo es 46XY. La RM muestra ausencia de surcos y bulbos olfatorios, hipoplasia del troncoencéfalo, platibasia e invaginación basilar.
El patólogo español, Maestre de San Juan, realizó en 1856 la primera descripción del SKMSJ en la autopsia de un varón con hipogenitalismo y ausencia de lóbulos olfatorios. En 1944 Kallmann describió la enfermedad como un síndrome genético en 3 familias2. Aunque la mayoría de los casos son esporádicos, aproximadamente un 20% son familiares. Se conocen 3 tipos de herencia3: ligada al cromosoma X, autosómica dominante y autosómica recesiva. Se han encontrado mutaciones en el gen KAL14, Xp22.31, encargado de la producción de anosmina, una proteína que facilita la migración neuronal desde la placoda olfatoria hasta el hipotálamo. Otras mutaciones descritas son las asociadas al receptor del factor de crecimiento de fibroblastos y a la proteína G acoplada al receptor 2 de la procineticina y a su ligando5.
La incidencia de este trastorno es muy variable (1 por cada 10.000 a 1 por cada 86.000) y es más frecuente en los varones5.
El diagnóstico se sospecha por presencia de hipogenitalismo, con concentraciones bajas de gonadotropinas y esteroides sexuales, que en la pubertad se normalizan tras la administración de Gn-RH. La anosmia se puede diagnosticar mediante anamnesis, pero la hiposmia requiere test olfatorios específicos. Como ocurrió en nuestros 2 pacientes, la RM6 contribuye a realizar el diagnóstico, puesto que detecta la ausencia o las alteraciones morfológicas de los bulbos olfatorios; sin embargo, hasta en el 25% de los pacientes puede ser normal, en estos casos los estudios genéticos son claves para el diagnóstico. Entre las malformaciones asociadas al SKMSJ, las que presenta el segundo paciente son muy frecuentes; sin embargo, la hernia diafragmática del primer caso es un hallazgo que no hemos encontrado descrito previamente.
El diagnóstico debe realizarse de forma precoz para que se pueda iniciar el tratamiento sustitutivo a una edad adecuada y mejorar así el estirón puberal y la mineralización ósea, aumentar la masa muscular y evitar posibles problemas de autoestima en adolescentes sin desarrollo de caracteres sexuales secundarios.
El tratamiento consiste en la inducción de la pubertad; se puede realizar mediante HCG por vía intramuscular administradas semanalmente, que facilitan la fertilidad pero, como sucedió en nuestro primer paciente, suelen ser mal toleradas debido a la frecuencia de las inyecciones. Los preparados de testosterona intramuscular de larga duración o en forma de gel son los tratamientos más cómodos y habituales.
