




Los síndromes monogénicos de insulinorresistencia sin lipodistrofia constituyen un grupo de entidades infrecuentes que incluyen los síndromes de Donohue o leprechaunismo, Rabson-Mendenhall y resistencia a la insulina tipo A. Se caracterizan por un amplio espectro fenotípico que asocia insulinorresistencia extrema y alteraciones hidrocarbonadas de grado variable.
Presentamos un caso de resistencia a la insulina tipo A, caracterizado por resistencia insulínica grave, acantosis nigricans e hiperandrogenismo, debido a una mutación en heterocigosis en el exón 19 del gen del receptor de insulina que codifica para el dominio tirosinquinasa.
Se destaca la elevada morbilidad de dicha entidad, a pesar de incluirse dentro del espectro menos grave de los síndromes genéticos de resistencia insulínica, así como la ausencia de una terapia satisfactoria. El estudio molecular revela el diagnóstico e informa del pronóstico y la supervivencia, factores ligados a la función residual del receptor, además de contribuir al desarrollo de nuevas dianas terapéuticas.
Insulin resistance syndromes without lipodystrophy are an infrequent and heterogeneous group of disorders with variable clinical phenotypes, associated with hyperglycemia and hyperinsulinemia. The three conditions related to mutations in the insulin receptor gene are leprechaunism or Donohue syndrome, Rabson-Mendenhall syndrome, and Type A syndrome.
A case is presented on a patient diagnosed with type A insulin resistance, defined by the triad of extreme insulin resistance, acanthosis nigricans, and hyperandrogenism, carrying a heterozygous mutation in exon 19 of the insulin receptor gene coding for its tyrosine kinase domain that is crucial for the catalytic activity of the receptor.
The molecular basis of the syndrome is reviewed, focusing on the structure-function relationships of the insulin receptor, knowing that the criteria for survival are linked to residual insulin receptor function. It is also pointed out that, although type A insulin resistance appears to represent a somewhat less severe condition, these patients have a high morbidity and their treatment is still unsatisfactory.
Los síndromes monogénicos de resistencia a la insulina constituyen un grupo de entidades infrecuentes, probablemente infradiagnosticadas en casos leves. Cursan con amplio espectro fenotípico, incluyendo hiperinsulinismo y resistencia insulínica severa, con grados diversos de alteración en el metabolismo de los carbohidratos1,2.
Se conocen 30 mutaciones en el gen del receptor de insulina (Ins-R) causantes de dichos síndromes (MIM 147670) con fenotipo variable, representando grados de un espectro continuo de gravedad, dependiendo de la disfunción del receptor: síndrome de insulinorresistencia tipo A (MIM 147670), caracterizado por resistencia extrema a la insulina, acantosis nigricans e hiperandrogenismo, sin asociar obesidad ni lipoatrofia3; síndrome de Donohue o Leprechaunismo (SD; MIM 246200), y el síndrome de Rabson-Mendenhall (SRM; MIM 262190)1-3.
Presentamos un caso de resistencia insulínica monogénica tipo A, enfatizando la importancia del estudio molecular, tanto para el mejor conocimiento de la patogénesis y la relación genotipo-fenotipo, como para la futura optimización del tratamiento. Destacamos la elevada morbilidad de este síndrome, a pesar de encuadrarse dentro del espectro más leve de los síndromes genéticos de resistencia extrema a la insulina.
Paciente y métodosAdolescente de 12 años y 8 meses, remitida desde Dermatología por hiperinsulinismo basal (231μU/ml). Natural de China y adoptada a los 9 meses de edad, desconociéndose antecedentes familiares y personales. Refería episodios frecuentes de debilidad, temblor, palidez y sensación de hambre cuando espaciaba la ingesta más de 3-4 h, con recuperación inmediata tras la ingesta, pero nunca se determinó la glucemia. No asociaba clínica cardinal de diabetes. Inició pubertad a los 10 años, con progresión normal de caracteres sexuales y sangrado vaginal único a los 11 años y 6 meses, con amenorrea secundaria desde entonces.
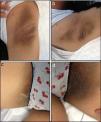
En el examen físico presentaba: talla de 160cm (+0,7 DE), peso de 58,8 Kg (+0,8 DE) e IMC de 22,9kg/m2 (+1,2 DE)4,5. Mostraba acantosis nigricans manifiesta en cuello, axilas e ingles (fig. 1), estadio Tanner con pubarquia 5, hipoplasia mamaria bilateral, más marcada en mama derecha (S3 derecha; S4 izquierda) y clitoromegalia (1,4cm de longitud). No se evidenció hirsutismo, hipertrofia muscular ni lesiones en la mucosa oral.
En la analítica mostraba glucemia basal de 95mg/dl, insulinemia basal de 266μUI/ml e hiperandrogenismo (tabla 1). La HbA1c y la glucemia en la sobrecarga oral de glucosa indicaron diabetes (tabla 1). El resto de las determinaciones analíticas fueron normales (tabla 1). La edad ósea fue de 14 años (Greulich & Pyle) y en la ecografía pélvica se objetivaron ovarios simétricos de tamaño aumentado (20 y 25 cc), junto con estroma central paucifolicular compatible con poliquistosis ovárica.
Determinaciones hormonales
| Basal | SOG (120 min) | |
| Glucemia (mg/dl) | 95/70 | 272 |
| Glucohemoglobina (VN %: 4,5-6,3) (VN IFCC: 25,7-45,4 mmol/mol) | 6,5 (IFCC: 47) | |
| Insulinemia (μUI/ml) (VN: 0-25,0) | 257/266 | 1.772 |
| CT (VN: 130,0-200,0 mg/dl) | 151 | |
| HDL-C (VN: 45,0-90,0 mg/dl) | 86 | |
| LDL-C (VN: 70,0-160,0 mg/dl) | 50 | |
| Triglicéridos (VN: 30-200 mg/dl) | 77 | |
| Testosterona total (VN: 14-76 ng/dl) | 349 | |
| Índice andrógenos libres (VN < 4,5) | 49,8 | |
| Δ4 androstendiona (VN: 0 4-3,3 ng/ml) | > 10 (0,4-3,3) | |
| 17 OHP progesterona (VN < 2 ng/ml) | 2,13 | |
| DHEAS (VN: 35-430 μg/dl) | 55.21 | |
| SHBG (VN: 18-114 nmol/l) | 24,3 | |
| FSH (mU/ml) y LH (mUI/ml); 17βEstradiol (pg/ml) | 2,7 y 6,4; 44 |
CT: colesterol total; FSH: hormona folículo-estimulante; HDL-C: colesterol unido a lipoproteínas de alta y baja densidad; LDL-C: colesterol unido a lipoproteínas de alta y baja densidad; LH: hormona luteinizante; SOG: sobrecarga oral de glucosa; VN: valor normal.
Se estudiaron en el ADN de leucocitos de sangre periférica6 los 22 exones del gen INS-R, utilizando cebadores específicos7-9, con secuenciación directa (3700 ABI). Se detectó una mutación, en heterocigosis, en el residuo 1131 (1158 si se incluye el péptido señal) del dominio tirosincinasa del exón 19 (fig. 2), ya descrita como patogénica en un paciente con SRM como heterocigoto compuesto, así como en un paciente con resistencia a la insulina tipo A descrita por Kishimoto et al.10,11. Al tratarse de una niña adoptada, carecemos de estudio genético de sus padres biológicos.
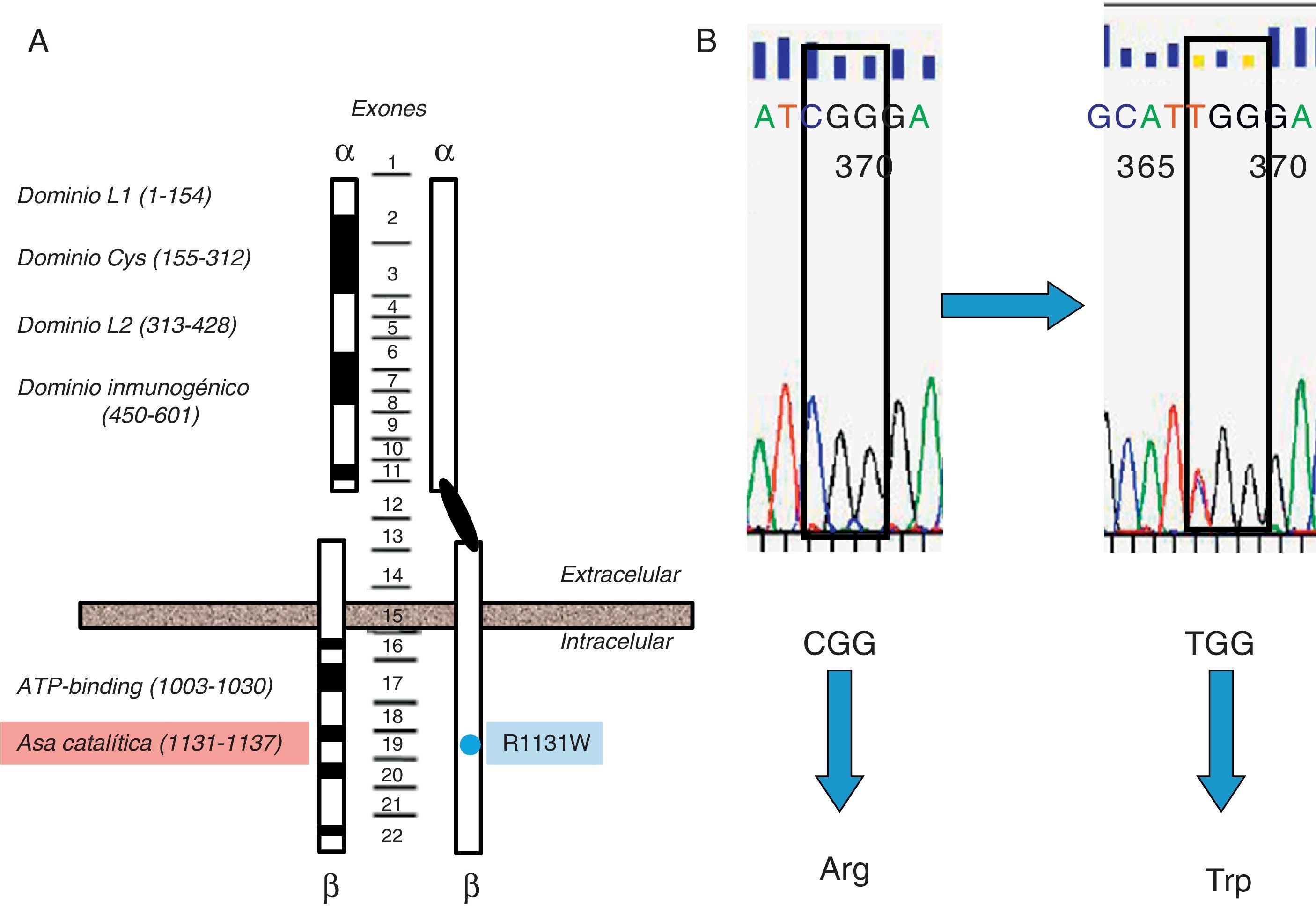
Representación esquemática del gen del receptor de insulina y mutación de la paciente. A) Representación esquemática del gen del receptor de insulina y localización de la mutación detectada en nuestra paciente (punto azul) afectando al dominio catalítico del receptor (Arg-Asp-Leu-Xaa1-Xaa2-Xaa3-Asn, residuos aminoácidos 1131-1137, localizados aproximadamente unos 100 residuos aguas abajo del sitio de unión a ATP) (sombreado en rojo). Los exones quedan indicados en el centro de la figura. B. Mutación Arg1131Trp de la paciente. B) Esferograma con la secuencia normal del gen del receptor de insulina (CGG→arginina) y la mutación en la paciente (TGG→triptófano).
Inició tratamiento con metformina (800mg/día por mala tolerancia a la dosis de 1.200mg/día) asociado a fraccionamiento de la ingesta de hidratos de carbono en 5 comidas, objetivándose regular control metabólico (HbA1c en torno a 6,2-6,3%), persistiendo hiperglucemias (hasta 240mg/dl a las 2 h tras la ingesta) y clínica ocasional de hipoglucemia (glucemia capilar de 48mg/dl). Recuperó la menstruación al mes y medio de tratamiento, con ciclos de 30-35 días.
DiscusiónEl Ins-R es una glucoproteína de superficie compuesta por 4 subunidades: 2 extracelulares (α) y 2 intracelulares (β). Cualquier defecto en su función puede ser responsable de síndromes de resistencia extrema a la insulina, como ocurre en este caso1,3.
Aunque desconocemos la prevalencia global de mutaciones del gen del Ins-R, algunos autores indican la posibilidad de estimación indirecta a partir de la incidencia de leprechaunismo, estableciendo un mínimo estimado de 1:1.000-1:2.000 para la frecuencia de alelos mutados en la población general de EE. UU.12.
Habitualmente, las mutaciones bialélicas del Ins-R humano se asocian típicamente a 2 síndromes de resistencia a la insulina: el SD, también conocido como leprechaunismo, y el SRM, ambos caracterizados por retraso de crecimiento intrauterino y posnatal, dismorfia facial, acantosis nigricans, hiperinsulinismo con hipoglucemia de ayuno e hiperglucemia posprandial.
El síndrome de IR tipo A se relaciona con mutaciones dominantes inactivadoras en el gen del Ins-R, localizadas en el dominio tirosincinasa del mismo, como la mutación en heterocigosis detectada en nuestro caso, aunque también están descritas mutaciones bialélicas1,13. No obstante, aunque el fenotipo clínico de los pacientes con síndromes monogénicos de resistencia a la insulina parece anticipar la diana del defecto molecular en el Ins-R, existe una considerable heterogeneidad. Así, en pacientes con IR tipo A existe variabilidad en el grado de resistencia a la insulina y evolución clínica, siendo aquellos con mutaciones en la subunidad α o mutaciones homocigóticas en la subunidad β los que tienden a tener fenotipos de mayor severidad. Por el contrario, mutaciones en heterocigosis en la subunidad β, como ocurre en nuestro caso, parecen predecir una menor severidad clínica1,9.
Aunque el genotipo en el locus del receptor de insulina parece ser el mayor determinante del fenotipo, no siempre explica las manifestaciones clínicas; se sospecha que factores ambientales (alimentación, ejercicio) influyan en la expresión clínica. Es probable que otros genes tengan también un impacto en la modulación de estos síndromes clínicos de resistencia extrema la insulina, como se ha indicado1,2.
La mutación Arg1131Trp, detectada en nuestro caso, afecta al primer residuo de arginina del bucle catalítico del Ins-R, residuo aminoácido universalmente conservado en todas la proteincinasas y crucial para su actividad catalítica (fig. 2)14. Dicha mutación, pero con distinto cambio de aminoácido (Gln1131; R1131Q), ha sido descrita por Kishimoto et al., confirmando su repercusión funcional y demostrando que es responsable de una disminución significativa de la capacidad de autofosforilación del receptor y, por tanto, de la actividad tirosincinasa10. Se han identificado diversas mutaciones en el bucle catalítico del Ins-R, alterando directamente la actividad tirosincinasa del receptor y siendo responsables de algunos de los síndromes monogénicos de insulinorresistencia en sus distintas expresiones9,15-18.
Las mutaciones en el dominio tirosincinasa inhiben también la endocitosis del receptor mutado, principal mecanismo de aclaramiento de insulina, lo que explica, parcialmente, el incremento de los niveles de insulina en estos pacientes. Además, la deficiente actividad tirosincinasa del receptor mutado explica la resistencia a la regulación a la baja del receptor inducida por la insulina, proceso que requiere igualmente la integridad del mecanismo de endocitosis1.
En relación con la aparente levedad del síndrome de IR tipo A, hay que destacar que la mayoría de los pacientes descritos en la literatura presentan diabetes en el momento de diagnóstico, como en el caso que describimos, asociando una alta morbimortalidad evolutivamente. Así, en el grupo de pacientes descrito por Musso et al., 6 de los 8 pacientes con IR tipo A tenían diabetes al diagnóstico y 7 de los 8 pacientes descritos presentaban diversos grados de retinopatía, nefropatía y/o neuropatía a los 30 años de evolución9. Aunque la mayoría de los casos asocian dislipidemia, nuestra paciente carecía de ella (tabla 1).
La mayoría de los pacientes mantienen un mal control metabólico a pesar del tratamiento9. Como la mayoría de los tratamientos disponibles son insatisfactorios, se han ensayado terapias alternativas (IGF-I o leptina recombinante humana) con resultados no concluyentes9,19,20.
En resumen, comunicamos un caso infrecuente de síndrome de insulinorresistencia severo tipo A, asociado a una mutación en el dominio tirosincinasa del Ins-R, crucial para su funcionalidad. Profundizar en el estudio molecular de los síndromes genéticos de resistencia insulínica es relevante, ya que el pronóstico y la supervivencia van unidos a la función residual del receptor, pudiendo permitir el desarrollo de nuevas dianas terapéuticas.
FinanciaciónCIBER de Fisiopatología de la Obesidad y Nutrición (CIBERobn), del Instituto de Salud Carlos III, Fondo de Investigación Sanitaria (FIS, PI09/91060 y PI10/00747), Fundación Endocrinología y Nutrición.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
