



La osteopetrosis (OP) es una rara enfermedad ósea congénita producida por una alteración funcional en los osteoclastos con incapacidad para la reabsorción ósea normal, produciéndose un aumento de la densidad mineral ósea y esclerosis ósea. Puede clasificarse en autosómica recesiva (OPTB) o autosómica dominante (OPTA1-2). Existe una gran variabilidad clínica de la enfermedad, desde asintomática a letal en los primeros meses de vida, con expresividad variable en los miembros de una familia. Su diagnóstico es principalmente clínico con confirmación genética, y el tratamiento es sintomático. Se presentan una serie de casos de OP, con el hallazgo de una nueva mutación en el gen LRP5 causante de OPTA1 en uno de ellos.
Osteopetrosis (OP) is a congenital bone disease which is caused by a functional disorder in osteoclasts with inability for normal bone resorption, leading to increased bone mineral density and bone sclerosis. It can be classified into different groups according to their clinical and their genetic characteristics: autosomal recessive with several subtypes (OPTB) or autosomal dominant type 1 or 2 (OPTA1-2). There is a wide clinical variability of the disease, from asymptomatic to lethal in the first months of life, with variable expressivity in the family members. Diagnosis is mainly clinical with genetic confirmation of the OP, and treatment is symptomatic. Three cases of OP are presented, with the discovery of a new gene mutation in LRP5 which caused OPTA1 in one of them.
La osteopetrosis (OP) se caracterizada por un incremento de la densidad ósea y esclerosis difusa del esqueleto. Estas alteraciones son consecuencia de un desequilibrio en el remodelado óseo, por un defecto funcional de los osteoclastos que les incapacita para la reabsorción ósea y cartilaginosa, formándose huesos más densos pero más frágiles1,2.
Existen distintos tipos de OP en función de su herencia (autosómica dominante [OPTA] o recesiva [OPTB]) y la clínica. Recientemente, se ha clasificado la osteopetrosis en OPTA tipo 1 y 2, y OPTB con varios subtipos, según la alteración genética que presentan. En función del subtipo de OPTB, la osteopetrosis puede resultar fatal en la primera infancia o ser lentamente progresiva con buen pronóstico. Por tanto, la OP presenta una gran variabilidad clínica: desde hallazgos casuales radiográficos en cráneo, vértebras o parrilla costal en pacientes asintomáticos, hasta fracturas espontáneas y complicaciones neurológicas derivadas de la compresión nerviosa por crecimiento óseo excesivo. Puede asociar alteraciones dentales como caries y osteomielitis, y acidosis tubular renal1–3.
El diagnóstico es principalmente clínico y radiográfico, confirmándose con estudio genético de la enfermedad. El tratamiento es sintomático, aunque recientemente se describe la realización de trasplantes medulares en los casos más severos.
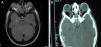
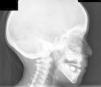
Casos clínicosEl caso 1 es un varón con gestación sin incidencias, parto a término con un peso de 2.890g, periodo neonatal, desarrollo ponderoestatural y psicomotor adecuados normales. Controlado en otorrinolaringología desde los 3 años por hipoacusia bilateral de transmisión con OMA de repetición. A los 511/12 años, acude al servicio de urgencias por proptosis ocular bilateral aguda tras episodio de vómitos de repetición y fiebre en contexto de cefalea de 2-3 semanas de evolución. En la exploración física y neurológica destaca exoftalmos bilateral y edema de papila bilateral. Se realizan radiografía y tomografía computarizada (TC) craneales, con aumento de densidad ósea generalizada, protrusión ocular izquierda con efecto masa intraorbitario dependiente del techo orbitario y elongación de ambos nervios ópticos. En la resonancia magnética (RM), se observan masas redondeadas intra y extraconales bilaterales de predominio izquierdo con efecto masa sobre músculo recto superior, diagnosticándose de encefalocele frontoorbitario izquierdo (fig. 1A y B). Como antecedentes familiares destaca el fenotipo materno: exoftalmos, prognatismo mandibular con ensanchamiento e hipoplasia malar.
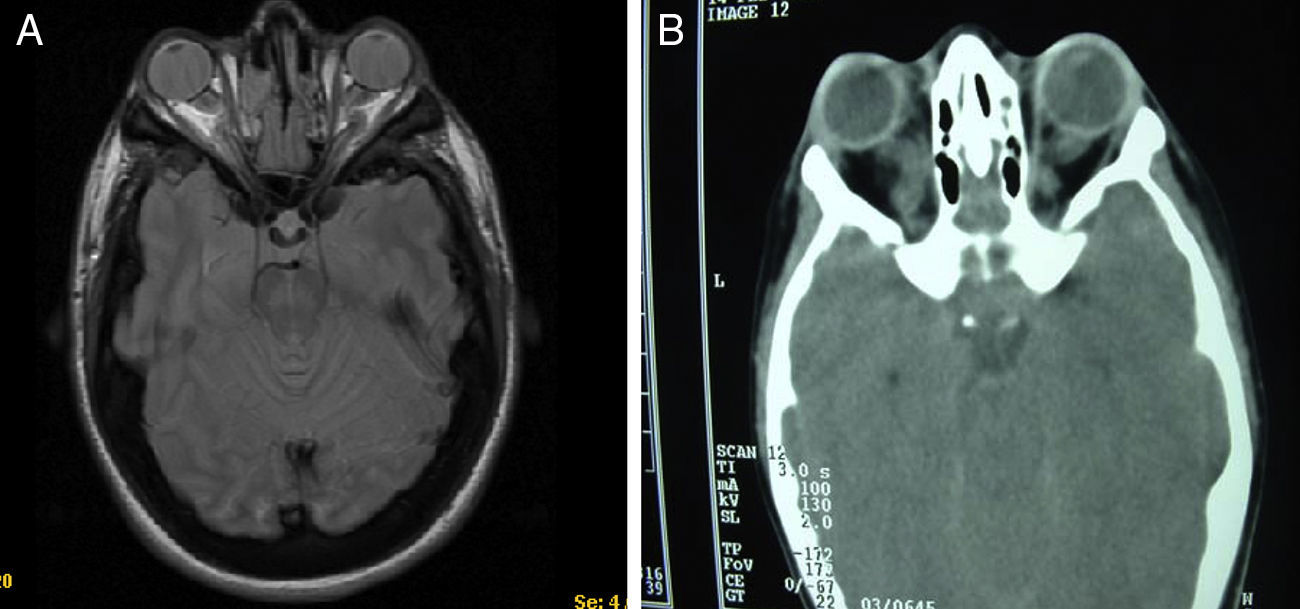
A y B) Imágenes de TAC y RM, en (A) se aprecia un aumento de densidad ósea generalizada, protrusión ocular izquierda, con efecto masa intraorbitario dependiente del techo orbitario y elongación de ambos nervios ópticos; y en (B) se aprecia masas redondeadas intra y extraconales bilaterales de predominio izquierdo, con efecto masa sobre músculo recto superior (encefalocele frontoorbitario izquierdo).
Se realiza densitometría ósea (DMO) por ultrasonidos a madre e hijo, siendo el Z-score de +3,51 y +2,89, respectivamente, confirmándose así la sospecha de osteopetrosis. A los 15 días reingresa por tortícolis, cefalea y envaramiento dorsal compatible con cuadro de hipertensión intracraneal, siendo remitido al hospital de referencia, donde es intervenido extirpando el encefalocele de manera exitosa. El estudio genético resultó negativo para el gen CLCN7, hallándose una mutación heterocigota en el exón 2 del gen LRP5 de la mutación c.335G>T;pGly112Val, compatible con osteopetrosis dominante. Su evolución ha sido satisfactoria, sin presentar clínica ni complicaciones. Actualmente, el paciente tiene 16 años, está asintomático y presenta una DMO +7,5, controlándose anualmente con RM, DMO y marcadores de recambio óseo.
El caso 2 es una niña de 59/12 años, de origen búlgaro, padres sanos no consanguíneos, embarazo controlado normal, parto eutócico a término con un peso de 3.030g y una longitud de 50cm, periodo neonatal, desarrollo psicomotor y ponderoestatural adecuados. Intervenida a los 4 años por mielogénesis imperfecta y policaries, precisando de varios implantes dentales. Controlada en oftalmología por epífora. A los 5 años, durante un episodio febril se objetiva en radiografía de tórax un aumento de densidad ósea generalizada mayor en esternón y columna vertebral. A la exploración física y neurológica destaca malposición dentaria con múltiples caries, siendo el resto normal. En la radiografía de cráneo aparece aumento de densidad ósea en calota, huesos de la base y cuerpos vertebrales cervicales, compatible con osteopetrosis (fig. 2). Ante la sospecha de alteración en el metabolismo óseo, se realiza bioquímica completa con marcadores de recambio óseos destacando un aumento de fosfatasa alcalina ósea (92,1U/l) y propéptido C-terminal de colágeno tipo I (231,1ng/ml), resto normal. En la DMO se aprecia un Z-score de +7,4, siendo normal en ambos padres. Estudio otorrinolaringológico y oftalmológico normales. Actualmente está pendiente de estudio genético.
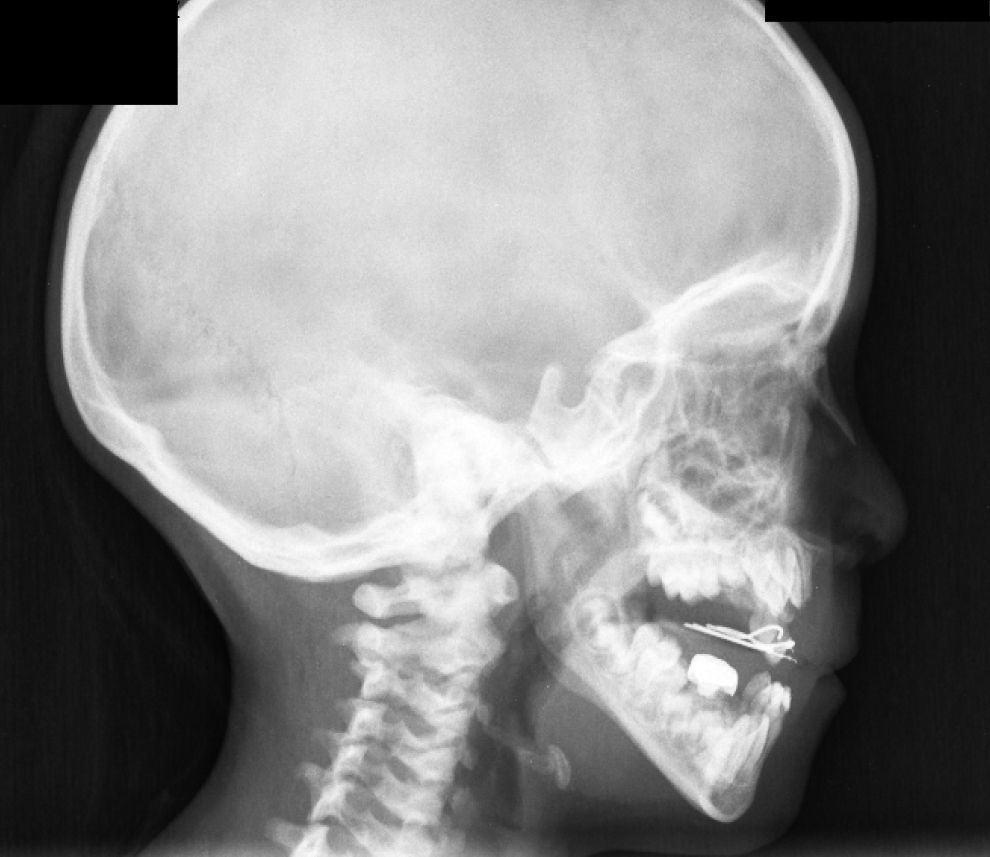
El caso 3 se trata de un paciente de 146/12 años, cuya madre y hermano presentan escoliosis leve. Embarazo y parto sin incidencias, con un peso de 3.570g y una longitud de 49cm, periodo neonatal sin alteraciones. Buen desarrollo ponderoestatural y psicomotor. En radiografía de columna (realizada para descartar escoliosis), presenta un aumento generalizado de la densidad mineral ósea en vértebras (fig. 3), costillas y huesos ilíacos. La exploración física normal, sin antecedentes de fracturas. Se realiza una DMO con Z-score +3,1. Entre los marcadores óseos realizados destaca fosfatasa alcalina ósea de 137,3U/l, osteocalcina de 113,2ng/ml, siendo el resto de analítica, orina y electroneurograma normales. Las densitometrías óseas por ultrasonidos realizadas a padres y hermano resultaron normales. Actualmente, el paciente está asintomático, con una DMO con Z-score de +2,7 y pendiente del estudio genético.
Discusión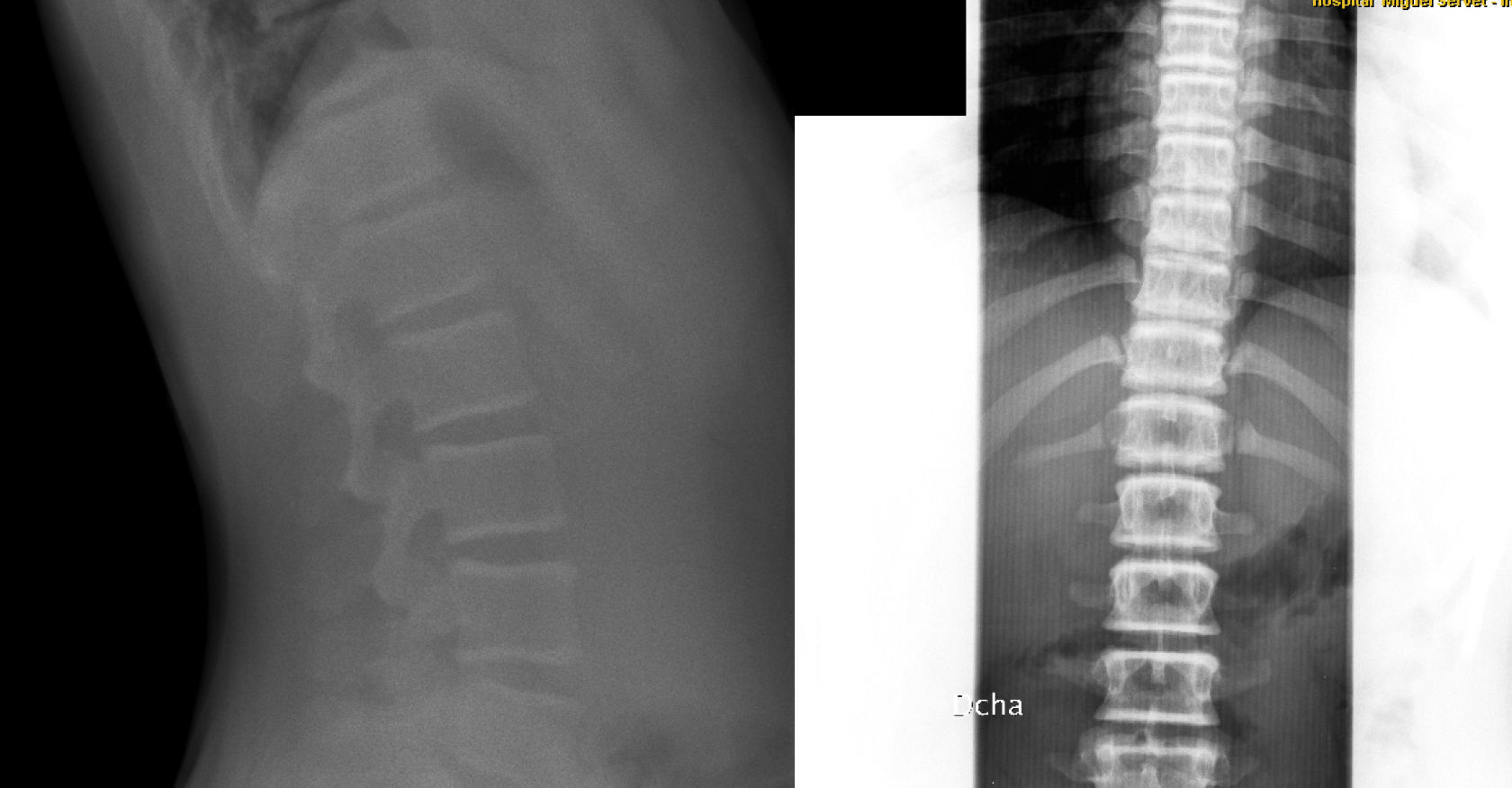
La osteopetrosis se produce por una alteración funcional en los osteoclastos, incapacitándolos para la reabsorción ósea normal. Existe una expresividad variable del proceso: diferentes formas clínicas y diferente severidad en la evolución de la enfermedad entre los familiares1,2. En la última década, son múltiples los estudios que investigan los diferentes genes implicados en el desarrollo de la OP. A su vez, el cromosoma 11q12-13 (que codifica varios genes relacionados con enfermedades óseas) ha sido una región prioritaria para analizar. En 2010, el Grupo de Nosología y Clasificación de las enfermedades esqueléticas genéticas, divide la OP AR en varios subtipos según su genética3,4: OPTB1-2-3-4-5-6-7; y la OP AD en 2 subtipos genéticos: OPTA1-2.
Clínicamente, la OPTA se caracteriza por esclerosis de la bóveda craneal en el tipo 1 o de la base craneal en el tipo 2, vértebras en «jersey de rugby» o «en sándwich», y alteraciones en la dentición (retraso en la erupción, malposición dentaria, caries)1,5. Como complicaciones, presentan alteraciones neurológicas por compresión nerviosa, disminución de la agudeza visual y de la audición, osteomielitis (mandibular la más típica), dolores osteoarticulares y fracturas patológicas (típicas en OPTA2). Es frecuente el diagnóstico casual en OPTA, o bien tras producirse fracturas patológicas o esclerosis en base del cráneo1,2. El paciente del caso 1, presenta una OPTA tipo 1. Los casos 2 y 3 presentados, quedan pendientes de su confirmación genética, siendo muy posible su diagnóstico de OPTA1 por su clínica asintomática y evolución lenta.
La clínica en la OPTB, además de los signos-síntomas típicos, viene definida por la genética: los genes TCIRG1 y CLCN7 (OPTB1 y 4, respectivamente) se asocian a la OP recesiva maligna, que junto con el RANKL (OPTB2), RANK (OPTB7) y OSTM1 (OPTB5), causan hasta el 70% de las OP AR malignas3,4,6,7. El gen que codifica la enzima anhidrasa carbónica II, se ha asociado a la OP recesiva con acidosis tubular renal o OPTB34,7,8. En el 2001, varios estudios empiezan a relacionar la OP AD autosómica dominante o OPTA2 con una mutación en el gen CLCN7 del cromosoma 16p13.37,9,10. También se empezó a asociar el gen low-density lipoprotein receptor-related protein (LRP5) como posible causante de la enfermedad en su forma dominante7,8.
El gen LRP5 se localiza en el cromosoma 11q12-13 y codifica una proteína expresada en los osteoblastos, cuyas mutaciones alteran la función de los mismos, disminuyendo o aumentando la densidad ósea (principalmente en cráneo y columna), asociándose respectivamente con el síndrome de osteoporosis-seudoglioma y síndromes con aumento de masa ósea, como es nuestro caso11–13. En 2002 Little et al.14 describen una mutación G171V en heterocigosis que produce un aumento de la masa ósea en los individuos portadores. Existen descritos 13 polimorfismos diferentes codificados en la región del gen y 6 mutaciones diferentes en los exones 2, 3 y 4 del gen LRP5, causantes de la OP AD (OPTA) en diversas familias12. Recientemente, se ha descrito la primera deleción monoalélica en el exón 3 del gen LRP5, con aumento de su función, causante de OP11. En el caso 1, se encontró una mutación en heterocigosis (c.335 G>T; pGly122Val) en el exón 2 del gen LRP5, no descrita en la literatura, y asociada al desarrollo de la OP AD.
El caso que se describe presenta una forma de presentación inusual y una nueva mutación no descrita hasta el momento, demostrando así la gran heterogeneidad del gen LRP5 en relación con enfermedades óseas al alterar su homeostasis, bien sea por aumento o por disminución de su función.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
