




Sr. Editor:
El síndrome hereditario de hiperferritinemia y cataratas (SHHC) se define por hiperferritinemia sin evidencia de sobrecarga férrica y cataratas congénitas como consecuencia de una mutación del IRE de caracter autosómico dominante1,2. Recientemente se han identificado algunas mutaciones de novo no familiares3,4, y es este caso el primero descrito en nuestro país.
Se trata de un varón de 16 meses de edad referido a consultas externas por hallazgo casual de hiperferritinemia en una analítica realizada por estudio de alergia a las proteínas de la leche de vaca (APLV). El niño se encontraba asintomático y salvo la APLV, no presentaba ningún antecedente personal relevante ni evidencia de enfermedad. No existían antecedentes familiares de hemocromatosis, cataratas congénitas o precoces ni otras alteraciones del metabolismo férrico. El paciente presentaba cifras elevadas seriadas de ferritina (1.454-1.489ng/ml) con patrón férrico normal: hierro, 86–147μg/dl; transferrina, 256–315mg/dl; capacidad total de transporte del hierro (TIBC), 325–400μg/dl; índice de saturación de la transferrina, 26,4-36,7%. La analítica realizada a los padres resultó rigurosamente normal (padre y madre con cifras de 236 y 32ng/ml de ferritina respectivamente). En la valoración oftalmológica se detectó una catarata con patrón en “miga de pan” compatible con depósitos de L-ferritina asociados al SHHC. El estudio genético demostró una mutación de novo en la posición 39 del IRE del gen de la L-ferritina (C39T), que no existía en los padres (figs. 1 y 2).
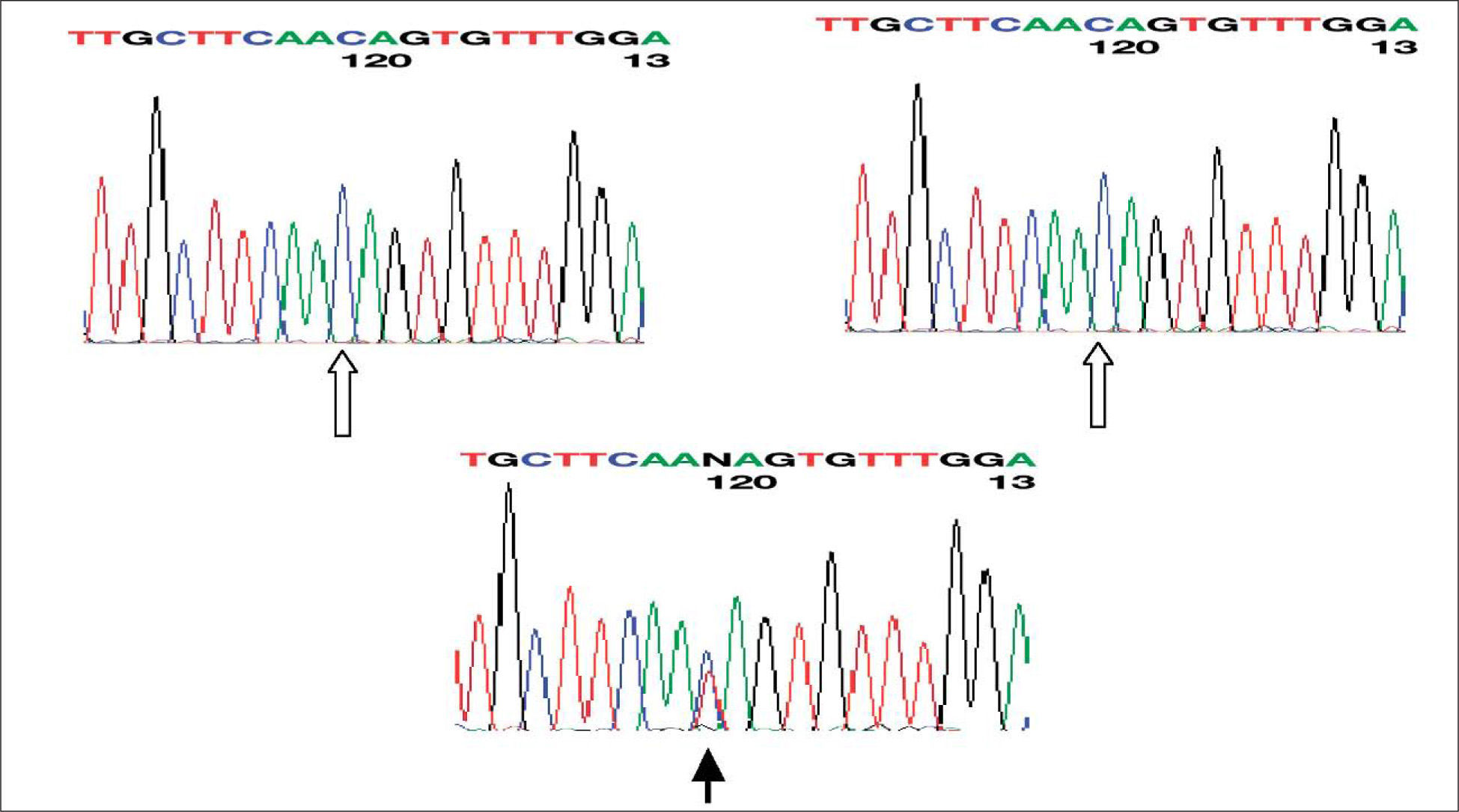
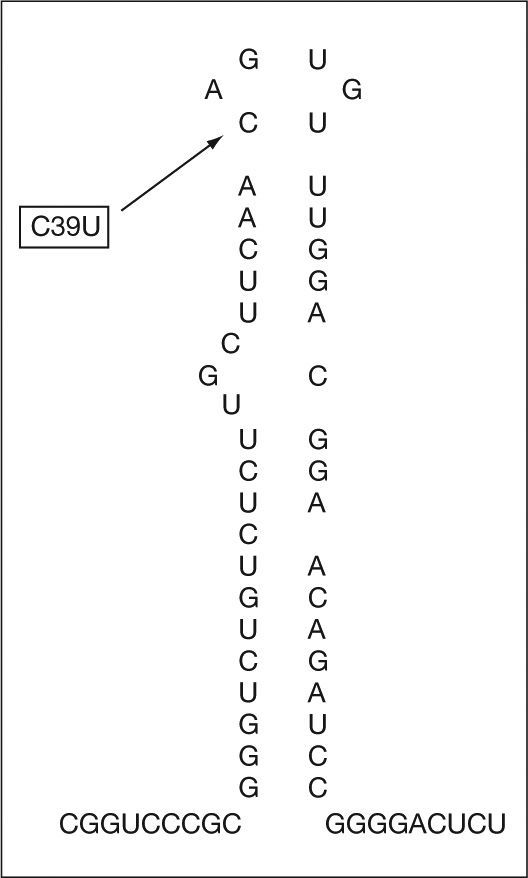
El SHHC es una entidad poco frecuente de reciente descripción1,2, pero posiblemente infraestimada por ser poco conocida. Se produce como consecuencia de una mutación del IRE en la región 5' no codificante del gen de la L-ferritina. Por este motivo disminuye la afinidad del IRE por la proteína IRP (iron regulatory protein). En condiciones normales cuando el IRE se une a la IRP (en situación de escasez de hierro) se inhibe la síntesis de ferritina. Sin embargo, en el SHHC esta retroalimentación negativa no funciona correctamente y existe por ello un exceso de ferritina en el organismo que es independiente de las concentraciones de hierro. Eso no produce otra patología en el organismo que cataratas nucleares por exceso de ferritina a ese nivel1,2,5.Se han descrito múltiples mutaciones del IRE, la mayoría esporádicas, y algunas delecciones. Cuando las mutaciones se localizan más cerca del bucle, las concentraciones de ferritina son mayores y más precoz la aparición de cataratas5, como ocurría en nuestro caso. En España se conocen varias familias afectadas6–9 –una de ellas descrita por los autores6– con diferentes mutaciones, pero no se había descrito ninguna mutación de novo. El SHHC es la causa más frecuente de hiperferritinemia con patrón férrico normal en pacientes aparentemente sanos, aunque las mutaciones de la ferroportina (hemocromatosis IV), también de transmisión autosómica dominante pueden presentar el mismo patrón analítico, pero con sobrecarga de hierro en el hígado4. Es importante conocer esta entidad, probablemente infradiagnosticada, para evitar exploraciones innecesarias encaminadas al diagnóstico de otros procesos con sobrecarga de hierro o realizar flebotomías, que podrían desencadenar anemia ferropénica.
