

El síndrome de Griscelli-Prunieras (SGP) es una rara entidad de herencia autonómica recesiva que se presenta con albinismo parcial. Su patogenia se explica por alteraciones en los genes que regulan el transporte de melanosomas. Se han descrito tres tipos en base a sus características genéticas y moleculares. Se conocen las mutaciones relacionadas con este síndrome. A continuación presentamos dos pacientes, no descritos anteriormente en España, que presentaban un cabello gris-plateado característico e inmunodeficiencia cuyo estudio genético demostró la mutación en el gen Rab27A (asociado al SG2). El pronóstico y tratamiento difiere considerablemente entre las diferentes formas de la enfermedad, por lo que el diagnóstico genético precoz es fundamental para poder establecer un pronóstico y tratamiento adecuados.
Griscelli-Prunieras syndrome (GS) is a rare autosomal recessive disorder characterized by partial albinism. His pathogenic mechanism is associated with defects in the packaging of melanin and other cellular proteins. GS is classified into 3 types based on the genetic and molecular features. Mutations in the genes which cause GS are known. We report two first cases described in Spain who presented a silver-gray sheen of the hair and a severe immune disorder. They were studied for mutations principally related to this syndrome. Two patients showed the Rab27a mutation (frequently associated with GS2). The natural disorder evolution differs considerably among the various forms, so a genetic study is essential in GS to achieve the most accurate prognosis and treatment possible.
El síndrome de Griscelli-Prunieras (SGP) es una enfermedad infrecuente, de herencia autosómica recesiva, que se manifiesta con albinismo parcial1. Se han descrito tres tipos de SGP entre la población del Mediterráneo Occidental, Turquía y dos casos recientes en América del sur2,3, según se acompañe de afectación neurológica (tipo 1), inmunológica (tipo 2) o albinismo de forma aislada (tipo 3). Todos los pacientes presentan hipopigmentación cutánea, fotosensibilidad, y un cabello gris-plateado característico. El pronóstico viene determinado por la afectación sistémica; así, los tipos 1 y 2 tienen una evolución fatal en la primera década de la vida, aunque el trasplante de médula ósea (TMO) puede ser curativo en este último4. Algunos pacientes han sobrevivido hasta la segunda década sin haber recibido terapia alguna, lo que sugiere la existencia de formas más leves de la enfermedad. Presentamos dos nuevos casos de SGP, enfermedad aún no descrita en España, y hacemos una revisión de esta entidad y su diagnóstico diferencial.
Casos clínicosCaso 1. Niña de 13 años de origen magrebí que consultó por tos productiva y fiebre de 39°C de 5 días de evolución. Refería cinco episodios de neumonía basal izquierda, dermatosis inespecíficas y úlceras bucales de repetición. Presentaba antecedentes familiares de consanguinidad (padres primos hermanos) y tres hermanos varones, con fenotipo similar al de la paciente, habían fallecido por cuadros febriles fulminantes no filiados. Al ingreso destacaba hipoventilación en base pulmonar izquierda, estancamiento pondoestatural (peso de 34kg y talla de 146cm, percentiles 5 y 10 para su edad y sexo, respectivamente) y albinismo parcial (coloración gris-plateada del cabello, cejas y pestañas) (fig. 1). La radiografía de tórax mostró un infiltrado-atelectasia en segmento postero-inferior del lóbulo pulmonar inferior izquierdo. Se inició antibioterapia oral con amoxicilina-clavulánico, quedando asintomática pero persistiendo la imagen radiológica a las 3 semanas. La tomografía computerizada torácica evidenció múltiples bronquiectasias en lóbulo inferior y língula del pulmón izquierdo. El estudio inmunológico mostró un déficit en la actividad de las células natural-killer (NK) y la respuesta de hipersensibilidad retardada. Las poblaciones linfocitarias, niveles de inmunoglobulinas y subtipos de inmunoglobulina G, la respuesta serológica a vacunas de virus atenuados y el test de nitroazul de tetrazolio fueron normales. En sangre periférica, no se observaron neutrófilos con gránulos lisosómicos gigantes. El estudio de la fibra capilar al microscopio óptico evidenció acúmulos largos e irregulares de melanina en forma de piqueteado, característicos de SGP. Se demostró la mutación homocigota IVS 3+3 A>G en el gen Rab27A (cromosoma 15q21), relacionada con el SGP tipo 2. La paciente ha requerido varios ingresos por infecciones respiratorias y una varicela grave. No ha desarrollado síndrome hemofagocítico (SHF) y no recibe ningún tratamiento específico, en espera de donante compatible para trasplante de médula ósea (TMO).

Caso 2. Niño de 3 años y 5 meses que consultó por fiebre de 40°C de un mes de evolución, adenopatías laterocervicales bilaterales y dolorosas, sangrado gingival y exantema cutáneo. No presentaba antecedentes personales de interés. De origen marroquí y con padres consanguíneos (primos hermanos). Tía materna fallecida a los 8 años por causa desconocida. A la exploración destacaban cabellos grisáceos y labios hiperpigmentados, un exantema micropetequial en extremidades inferiores y tronco, hepato y esplenomegalia y pequeñas adenopatías laterocervicales bilaterales, móviles y elásticas. La analítica sanguínea mostró plaquetopenia de 11.500/μL, leucopenia (4000/μL con desviación a la izquierda), PCR elevada (118mg/l) y hipertransaminasemia (GOT 450UI/l, y GTP 153UI/l), Paul-Bunnell negativo. La ecografía abdominal objetivó una hepatoesplenomegalia con vesícula biliar hidrópica. Se recogieron cultivos de sangre y orina, se realizaron estudios microbiológicos y se inició empíricamente antibioterapia con cefotaxima endovenosa. Se confirmó la infección por virus de Epstein-Barr (por reacción en cadena de la polimerasa plasmática), iniciándose aciclovir. Ante la persistencia del cuadro clínico y los hallazgos analíticos descritos se realizó biopsia de médula ósea que fue compatible con síndrome hemofagocítico (SHF). Se inició el protocolo terapéutico HLH 20045 (dexametasona, ciclosporina y etopósido), con progresiva normalización clínica y analítica. El estudio inmunológico mostró un déficit en la actividad de las células NK y en la respuesta de hipersensibilidad retardada. Como en el caso anterior, los cabellos gris-plateados, al microscopio óptico, mostraban acúmulos atípicos de melanina. Se objetivó la delección homocigota en el gen Rab27A en los exones 3 y 4 (cromosoma 15q21) confirmando el diagnóstico de SGP tipo 2. Actualmente sigue en tratamiento quimioterápico.
En ambos casos se cursó estudio en padres y hermanos, que resultaron heterocigotos para alguna de las mutaciones descritas, y se realizó consejo genético a la familia.
DiscusiónEn 1978, Griscelli y Prunieras1 describieron un síndrome autosómico recesivo asociado a albinismo parcial e inmunodeficiencia, clínicamente muy similar al síndrome de Chediak-Higashi. Los avances en las técnicas de diagnóstico genético-molecular han permitido dotar a esta patología de entidad propia, conociéndose desde 19942 como el SGP. Se han publicado unos 76 casos de SGP (4 casos, 70 casos y 2 casos de los tipos 1, 2 y 3, respectivamente)6,7. La consanguinidad es una característica común en la mayoría de pacientes descritos.
Se clasifica en tres tipos en base a las características clínicas, genéticas y moleculares8,9. El SGP tipo 1 es causado por una mutación en el gen que codifica la miosina Va (MYO5A), localizado en el cromosoma 15q21, regulador del transporte de melanina a las organelas de los melanocitos y neuronas10–13. Cursa con albinismo parcial y trastornos neurológicos causados por el depósito patológico de melanina en el tejido neuronal. La resonancia magnética puede mostrar atrofia cerebelar congénita14. El SGP tipo 2, que corresponde a los pacientes presentados, fue descrito por Harfi15. Está causado por la mutación del gen Rab27A, localizado también en el cromosoma 15q21 y que codifica para una GTPasa que interviene en el transporte de melanina desde los melanocitos a los queratinocitos, y en la correcta degranulación citotóxica de los linfocitos NK. Los portadores homocigotos de esta mutación muestran un defecto en la función citotóxica de los linfocitos NK y en la respuesta de hipersensibilidad retardada, presentando infecciones bacterianas de repetición y SHF16, a menudo desencadenado por infecciones previas, bacterianas o víricas, como en el caso que se presenta9. Las manifestaciones neurológicas del SHF pueden ser el primer signo clínico y son reversibles con la remisión del cuadro, a diferencia de los síntomas neurológicos del SGP tipo 1, presentes desde el nacimiento, estables e irreversibles. Recientemente, se ha descrito un el SGP tipo 3, que representa la forma más benigna de la enfermedad11 y que se caracteriza por albinismo parcial sin afectación inmune ni neurológica acompañantes. Se han identificado dos mutaciones distintas en los dos casos descritos hasta la fecha (gen MLPH en el cromosoma 2q37.3 y gen MYO5A en el cromosoma 15q21)17.
La edad al diagnóstico del SGP es variable: precoz por la sintomatología neurológica irreversible en el SGP tipo 1 y en edad escolar, o quizás infradiagnosticado, en el SGP tipo 3. En el SGP tipo 2 se han descrito casos fulminantes en el periodo neonatal por SHF, aunque la media de edad al diagnóstico es de 17 meses13.
El estudio inmunológico de los pacientes con SGP tipo 2 revela un fallo en la función NK, con quimioluminiscencia, quimiotaxis y actividad bactericida disminuidas. El test de nitroazul de tetrazolio no se altera, sugiriendo que la actividad oxidativa y la producción de radicales superóxido están conservadas. Presentan ausencia en la respuesta de hipersensibilidad retardada e hipogammaglobulinemia a expensas de los niveles plasmáticos de inmunoglobulinas G y A11.
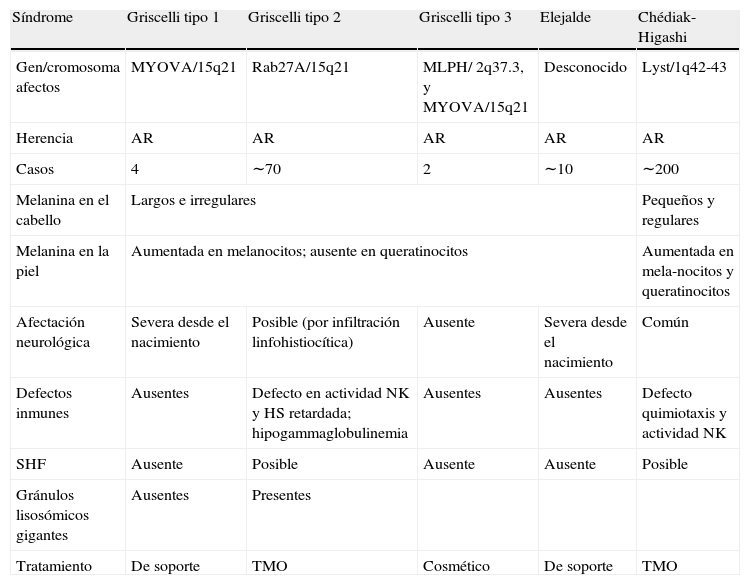
En todas las formas, el estudio del cabello al microscopio óptico muestra agregados largos e irregulares de melanina en la médula de la fibra capilar. La biopsia cutánea revela melanocitos cargados de melanina y escaso pigmento en los queratinocitos adyacentes. El diagnóstico definitivo lo da el estudio genético-molecular de la mutación, que permite ofrecer consejo genético. Debe plantearse el diagnóstico diferencial del SGP con los síndromes de Chediak-Higashi y Elejalde18 (tabla 1).
Diagnóstico diferencial de las patologías que cursan con albinismo parcial e inmunodeficiencia
| Síndrome | Griscelli tipo 1 | Griscelli tipo 2 | Griscelli tipo 3 | Elejalde | Chédiak-Higashi |
| Gen/cromosoma afectos | MYOVA/15q21 | Rab27A/15q21 | MLPH/ 2q37.3, y MYOVA/15q21 | Desconocido | Lyst/1q42-43 |
| Herencia | AR | AR | AR | AR | AR |
| Casos | 4 | ∼70 | 2 | ∼10 | ∼200 |
| Melanina en el cabello | Largos e irregulares | Pequeños y regulares | |||
| Melanina en la piel | Aumentada en melanocitos; ausente en queratinocitos | Aumentada en mela-nocitos y queratinocitos | |||
| Afectación neurológica | Severa desde el nacimiento | Posible (por infiltración linfohistiocítica) | Ausente | Severa desde el nacimiento | Común |
| Defectos inmunes | Ausentes | Defecto en actividad NK y HS retardada; hipogammaglobulinemia | Ausentes | Ausentes | Defecto quimiotaxis y actividad NK |
| SHF | Ausente | Posible | Ausente | Ausente | Posible |
| Gránulos lisosómicos gigantes | Ausentes | Presentes | |||
| Tratamiento | De soporte | TMO | Cosmético | De soporte | TMO |
AR, autosómica recesiva; NK, natural killer; HS, hipersensibilidad retardada; SHF, síndrome hemofagocítico; TMO, trasplante de médula ósea.
El tratamiento en el SGP tipo 1 es sintomático y de soporte. En el SGP tipo 2, el TMO es curativo, y se recomienda realizarlo precozmente. Cuando se desarrolla SHF, el uso de inmunosupresores y altas dosis de corticoides logra remisiones, en espera de donante, aunque no se ha mostrado eficaz en el control de las recidivas19,20. En el SGP tipo 3, no se requiere más intervención que el consejo dermatológico.
En conclusión, el SGP es una entidad infrecuente y con morbimortalidad elevadas cuando asocia manifestaciones sistémicas. Presenta una distribución geográfica específica, pero cabe tenerlo presente como enfermedad importada. La correlación entre el genotipo y la evolución natural de la enfermedad viene determinada por el tipo de mutación. Ante el fenotipo descrito es necesario un alto grado de sospecha clínica inicial para llegar al diagnóstico genético y poder plantear precozmente la posibilidad de un TMO.
