




Los síntomas/signos indicativos de una coagulopatía son un motivo de consulta frecuente en las unidades de Hematología Pediátrica. Tanto la clínica como los antecedentes familiares son fundamentales para el diagnóstico.
Pacientes y métodosEstudio retrospectivo y descriptivo de los pacientes derivados a una consulta de Hematología Pediátrica de un hospital de tercer nivel por posible coagulopatía durante el año 2012.
ResultadosSe estudiaron 47 niños. El 61,7% no había presentado previamente sangrado. El motivo de derivación más frecuente fue un tiempo de tromboplastina parcial activada alargado sin hemorragia (42,5%); de estos, un 25% fue diagnosticado de una coagulopatía con riesgo real de sangrado. En los pacientes derivados por tiempo de tromboplastina parcial activada alargado con clínica hemorrágica se detecta una coagulopatía con riesgo real de sangrado con mayor frecuencia (41,7%). En los niños con antecedentes familiares de sangrado se diagnostica con más frecuencia una coagulopatía con riesgo real de sangrado: 37,5 vs. 14,3% (niños sin antecedentes familiares). Los diagnósticos han sido: sano (48,9%), enfermedad de von Willebrand tipo1 (19,1%), déficit de factor xii (19,1%), déficit de factor xi (4,2%), déficit de precalicreína/cininógeno de alto peso molecular (2,1%), déficit adquirido de factor x (2,1%) y déficit de factor ix (2,1%).
ConclusionesLos antecedentes personales y familiares de sangrado orientan el diagnóstico de una coagulopatía. El motivo de derivación debería basarse en mayor medida en la clínica hemorrágica y no solo en un tiempo de laboratorio alterado. Los diagnósticos más frecuentes han sido enfermedad de von Willebrand tipo 1 y déficit de factor xii.
Symptoms/signs suggestive of coagulopathy is a frequent complaint in Pediatric Hematology units. Both the clinical and family history are essential for diagnosis.
Patients and methodsRetrospective and descriptive study of patients referred to a Pediatric Hematology unit of a tertiary hospital for possible coagulopathy during 2012.
ResultsA total of 47 children were studied, of whom 61.7% had not previously suffered bleeding. The most frequent reason for referral was an eloganted activated partial thromboplastin time without any hemorrhage (42.5%), of these, 25% were diagnosed of a coagulopathy with a real risk of bleeding. While patients referred due to an eloganted activated partial thromboplastin time with bleeding more frequently (41.7%) have a coagulopathy with a real risk of bleeding. Children with a family history of bleeding are diagnosed more frequently with a coagulopathy with a real risk of bleeding: 37.5% (family history) vs. 14.3% (without). The most frequent diagnoses were: healthy children (48.9%), von Willebrand type 1 disease (19.1%), factor xii deficiency (19.1%), factor xi deficiency (4.2%), prekalikrein/high molecular weight kininogen deficiency (2.1%), acquired deficiency of factor x (2.1%), and factor ix deficiency (2.1%).
ConclusionsA thorough personal and family bleeding history and physical examination are the first steps for a correct differential diagnosis. The reason for referral should be based more on clinical bleeding and not just on an abnormal coagulation time. The most frequent diagnoses were type 1 von Willebrand disease and factor xii deficiency.
Los signos indicativos de una coagulopatía son un motivo de consulta frecuente en Hematología. Su diagnóstico puede ser difícil debido a los patrones de sangrado específicos de la edad: la epistaxis (39%) y los hematomas de aparición fácil (24%) son frecuentes sin ningún trastorno hemorrágico. Pero a su vez estos pueden ser la única clínica reportada en niños con una coagulopatía que no han sido sometidos a ningún desafío hemostático1.
Además, el diagnóstico diferencial de sangrado/hematomas en la infancia incluye otras entidades, como: alteraciones plaquetarias, vasculopatías y maltratos.
Entre las coagulopatías hereditarias, la más frecuente es la enfermedad de von Willebrand (evW). Su prevalencia es aproximadamente de un 1%, según estudios clásicos2,3. Pero estudios más recientes hablan de una prevalencia de 0,1% de evW clínicamente sintomática en niños4. Le seguiría en frecuencia el déficit de factor viii (hemofilia A) y el déficit de factor ix (hemofilia B), con una prevalencia de 1:5.000 varones y 1:30.000 varones, respectivamente5.
Existen déficits de factores que no se traducen en sangrados, como el déficit de alguno de los factores de contacto: cininógeno de alto peso molecular (HMWK), precalicreína y factor xii. Estos factores no intervienen en la fase de propagación de la cascada de la coagulación y, por tanto, su déficit, a pesar de alargar in vitro el tiempo de tromboplastina parcial activada (TTPa), no se asocia a sangrado in vivo6.
Con el presente estudio se pretende conocer los servicios que más pacientes remiten para estudio de coagulación, el motivo de la derivación, relacionar los datos de laboratorio con la clínica de sangrado y saber los diagnósticos más frecuentes en nuestro entorno. Un mejor conocimiento de estos pacientes debe servir para mejorar el circuito diagnóstico.
Pacientes y métodosEstudio retrospectivo y descriptivo de los niños estudiados por posible coagulopatía en la Unidad de Onco-Hematología pediátrica del HUV Macarena de Sevilla durante el año 2012.
En esta unidad, en el año 2012, se estudiaban y trataban todos los tumores sólidos/hematológicos de la infancia (excepto los tumores cerebrales) y la hematología no oncológica. Los pacientes diagnosticados de hemofilia A o B eran remitidos tras el diagnóstico a la Unidad de Hemofilia del HUV Rocío de Sevilla.
En 2012 hubo 20 pacientes nuevos oncológicos, y 120 hematológicos.
La clínica es llevada por pediatras hematólogos y las técnicas de laboratorio por hematólogos de adultos.
Se han tenido en cuenta los niños con: algún tiempo de coagulación alterado, aquellos con tiempos de coagulación normales y sangrado (tras descartar la trombocitopenia) y aquellos estudiados por tener un familiar con alguna coagulopatía hereditaria. Se han estudiado un total de 52 niños. De estos, 5 eran pacientes oncológicos en tratamiento activo y se han descartado por las posibles interacciones de la quimioterapia y de la enfermedad de base.
Se han tenido en cuenta finalmente los 47 niños restantes.
El diagnóstico de cualquier coagulopatía se ha realizado con un nivel plasmático del factor inferior a 45%. En el caso de la evW1, el cofactor de la ristocetina tenía que ser también inferior al 45%, y presentar una clínica/antecedentes familiares (AF) compatibles.
Se ha considerado el déficit del factor xii, precalicreína y HMWK como déficits sin riesgo real de sangrado.
Se analizan las siguientes variables: edad, sexo, servicio que deriva al paciente, motivo de la derivación, AF de coagulopatía/sangrado, clínica de sangrado previa y localización de este, número de analíticas realizadas y diagnóstico final.
Se ha considerado una historia familiar positiva si existía un sangrado cutáneo y/o mucoso significativo en un familiar de primer grado. No se han utilizado cuestionarios estandarizados de sangrado.
Todos los datos obtenidos y considerados como variables a estudio fueron almacenados y estudiados con el paquete de software estadístico SPSS® v.15.0 (SPSS Inc., Chicago, IL. EE. UU.).
Análisis estadístico descriptivo: las variables cualitativas se expresan como frecuencias absolutas y porcentajes. Las variables cuantitativas, como media, mediana y rango.
Análisis estadístico analítico: se ha utilizado el test exacto de Fisher para comparar frecuencias de 2 variables cualitativas, el test Chi-cuadrado de bondad de ajuste para comparar frecuencias de una misma variable cualitativa, y la prueba de Chi cuadrado de Pearson para examinar la relación entre 2 variables cualitativas. Se consideró un nivel de significación p<0,05.
ResultadosSe han recogido los datos de un total de 47 niños: 28 niños (59,6%) y 19 niñas (40,4%).
La edad media en la primera visita fue de 6,1 años (mediana: 6 años; intervalo: 1 mes-13 años).
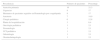
La procedencia de los pacientes se recoge en la tabla 1.
Procedencia de los niños estudiados por coagulopatía
| Procedencia | Número de pacientes | Porcentaje |
|---|---|---|
| Atención primaria | 10 | 21,3 |
| Urgencias | 8 | 17 |
| Hermanos de pacientes seguidos en Hematología por coagulopatía | 8 | 17 |
| Anestesia | 7 | 14,9 |
| Cirugía pediátrica | 6 | 12,8 |
| Planta de hospitalización | 3 | 6,4 |
| Oncología pediátrica | 1 | 2,1 |
| Neonatología | 1 | 2,1 |
| UCI pediátrica | 1 | 2,1 |
| Odontología | 1 | 2,1 |
| Otorrinolaringología | 1 | 2,1 |
La media de analíticas realizadas para llegar al diagnóstico fue de 2 (mediana: 2; intervalo: 1-5).
- •
Clínica hemorrágica y diagnósticos:
De los 47 pacientes, 18 (38,3%) habían presentado previamente algún sangrado, siendo la epistaxis (12 niños, 66,6%) la clase más frecuente.
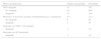
En un niño no se llegó al diagnóstico porque la familia dejó de acudir a las citas. Se resumen en la tabla 2 los diagnósticos finales y la presencia/ausencia de sangrados y si existen diferencias estadísticamente significativas entre sangrado/no sangrado dentro de cada grupo diagnóstico.
- •
AF de sangrado:
Diagnósticos finales y presencia/ausencia de clínica hemorrágica
| Diagnóstico | Sangrado | Número de pacientes | Porcentaje | pa |
|---|---|---|---|---|
| Sano | 23 | 48,9 | ||
| Sí | 7 | 30,4 | 0,061 | |
| No | 16 | 69,6 | ||
| EVW1 | 9 | 19,1 | ||
| Sí | 6 | 66,7 | 0,058 | |
| No | 3 | 33,3 | ||
| Déficit de factor XII | 9 | 19,1 | ||
| Sí | 3 | 33,3 | 0,317 | |
| No | 6 | 66,7 | ||
| Déficit de factor XI | No | 2 | 4,2 | |
| Déficit de factores de contacto (diferentes al XII) | No | 1 | 2,1 | |
| Déficit de factor X adquirido | Sí | 1 | 2,1 | |
| Déficit de factor IX | No | 1 | 2,1 | |
| Perdidos | Sí | 1 | 2,1 |
En 2 pacientes no se pudieron recoger por ser adoptados.
En la tabla 3 se resume la presencia/ausencia de AF de sangrado según el diagnóstico final. En los niños diagnosticados de una coagulopatía sin riesgo real de sangrado se recogen con menor frecuencia AF de sangrado (p=0,029).
- •
Motivo de la derivación:
Antecedentes familiares de sangrado
| Diagnóstico final | Total (n=45) | Con AF de sangrado (n=24) | Sin AF de sangrado (n=21) | p* |
|---|---|---|---|---|
| Sanos | 23 (51,1) | 13 (56,5) | 10 (43,5) | NSa |
| Coagulopatía sin riesgo de sangrado | 10 (22,1) | 2 (20) | 8 (80) | 0,029a |
| Coagulopatía con riesgo de sangrado | 12 (26,7) | 9 (75) | 3 (25) | 0,07a |
AF: antecedentes familiares; NS: diferencias estadísticamente no significativas.
Datos expresados como n (%).
Se resume en la tabla 4.
Motivo de derivación
| Motivo de derivación | Número de pacientes | Porcentaje |
|---|---|---|
| TTPa alargado | 33 | 70,2 |
| No sangrado | 20 | 66,6 |
| Sangrado | 13 | 39,4 |
| Hermanos de pacientes seguidos en Hematología por coagulopatía | 10 | 21,3 |
| No sangrado | 9 | 90 |
| Sangrado | 1 | 10 |
| Sangrado con TTPa y AP normales | ||
| Sangrado | 2 | 4,3 |
| Sangrado con AP disminuida | ||
| Sangrado | 2 | 4,3 |
AP: actividad de protrombina; TTPa: tiempo de tromboplastina parcial activado.
Se analiza a continuación cada grupo:
- -
TTPa alargado sin sangrado: 20 niños. El diagnóstico final fue: 8 (40%) niños sanos, 6 (30%) con déficit de factor xii (uno de ellos se diagnostica, además, de la presencia de un anticoagulante lúpico en el contexto de sepsis), 3 (15%) con evW1, uno (5%) con déficit de factor xi, otro (5%) con déficit de otros factores de contacto (HMWK, precalicreína) y un último (5%) con déficit de factor ix.
- -
TTPa alargado con sangrado: 13 niños, aunque en uno de ellos se perdió el seguimiento. Se resumen a continuación el tipo de sangrado y el diagnóstico final:
- a)
Epistaxis: 10 (77%). El diagnóstico final fue: 5 pacientes (50%) con evW1 (uno de ellos se diagnosticó, además, de un anticoagulante lúpico); 2 pacientes (20%) con déficit de factor xii; dos pacientes (20%) sanos.
- b)
Hemorragia a otro nivel: 3 (23,1%). El diagnóstico final fue: 2 niños sanos (66,7%), que habían sido referidos por hematemesis en un caso y petequias en el segundo, y un paciente con déficit de factor xii (33,3%), que había presentado hemorragia intraventricular en el contexto de prematuridad.
- a)
Excluyendo al paciente sin seguimiento y al prematuro, 5 (45,5%) de los 11 pacientes derivados por TTPa alterado y sangrado se diagnosticaron de una coagulopatía con riesgo real de sangrado. Por otra parte, en el grupo de niños con TTPa alargado sin clínica de sangrado, 5 de los 20 (25%) se diagnosticaron de alguna coagulopatía con riesgo real de sangrado. Pero estas diferencias no son estadísticamente significativas.
- -
Trastorno hemorrágico familiar: 10 niños. Los diagnósticos finales fueron:
- a)
Sanos: 9 (90%). Ocho sin sangrado y uno con epistaxis ocasional.
- b)
Déficit de factor xi: un paciente (10%) sin sangrado.
- a)
- -
Sangrado con TTPa y actividad de protrombina (AP) normales: un niño fue diagnosticado de evW1 tras una hemorragia en sábana tras intervención de fimosis. Otro niño, remitido por epistaxis y hemorragia tras extracción dentaria, no se diagnosticó de ninguna alteración.
- -
Sangrado con AP disminuida: un niño con TTPa también alargado con clínica de rectorragia fue diagnosticado de déficit de factor x secundario a un déficit de vitamina K. Otro niño, con hematomas de repetición, no se diagnosticó de ninguna alteración.
Los resultados del estudio muestran que, en nuestra unidad de Hematología Pediátrica, la mayoría de los niños remitidos para descartar una coagulopatía son derivados por un TTPa alargado sin clínica hemorrágica, y que el 75% de estos y un 48,7% del total de niños estudiados son sanos.
¿Estaba justificada la derivación? ¿Es necesario hacer un estudio de coagulación a aquellos pacientes sin historia de sangrado? Los datos evidencian que seguramente se deberían mejorar los pasos previos a la derivación a Hematología. ¿Cuándo estaría indicado hacer un estudio de coagulación básico? ¿Y a quiénes de los que tuviesen algún tiempo alterado se les debe ampliar el estudio? Las derivaciones procedentes de Cirugía y Anestesia, sumadas, representan el motivo de consulta más frecuente: previamente a cualquier tipo de cirugía se hace sistemáticamente, sin tener en cuenta ni la historia personal de sangrado ni los AF, un TTPa y una AP.
Pero la American Academy of Otolaryngology-Head and Neck Surgery recomienda que el estudio de coagulación preintervención solo se realice si por la historia personal y/o familiar se sospecha una coagulopatía7. Del mismo modo, el British Committee for Standards in Hematology recomendó en 2008 no hacer un cribado universal de la coagulación previa a las cirugías, sino obtener una historia de sangrado completa8. Cooper et al. indican que la estrategia más rentable sería no realizar estudios de coagulación previos a una amigdalectomía en niños9. Siguiendo con la amigdalectomía, una de las intervenciones más frecuentes en niños, Zagólski detectó sangrado tras amigdalectomía en varios pacientes con pruebas de coagulación previas normales, sin objetivarse sangrado en pacientes con TTPa alterado previamente. Estos autores apuntan que la incidencia de hemorragia postamigdalectomía no aumenta en los pacientes con TTPa alargados, y que lo realmente relevante para predecir el riesgo de sangrado tras una amigdalectomía son los antecedentes personales de epistaxis recurrentes, hematomas tras traumatismos menores y hemorragia prolongada tras lesiones menores10. En el estudio de Hubner se concluye que la historia cuidadosa y dirigida resulta de alto valor predictivo en el diagnóstico de las coagulopatías. La ausencia de sangrados permite predecir que los resultados de laboratorio serán normales en alrededor de un 94% de los casos11.
Pero, por otro lado, ¿cómo se diagnosticarían aquellas coagulopatías que no han cursado aún con hemorragia si no se realizan estudios de laboratorio? Según Bowman et al., la ausencia de sangrado en un niño con evW puede ser debida a la falta de intervenciones quirúrgicas, extracciones dentarias o menstruaciones que hayan puesto a prueba la coagulación4. Es entonces cuando los AF juegan un papel crucial en la orientación diagnóstica. Es más, el diagnóstico de ciertas coagulopatías, como la evW, requiere la presencia de unos AF positivos12.
El diseño de nuestro estudio es limitado para obtener conclusiones en cuanto a los AF de sangrado. Únicamente se puede comentar que el grupo de niños diagnosticados de una coagulopatía con riesgo real de sangrado es el grupo que con más frecuencia tiene AF positivos.
El número medio de analíticas realizadas ha sido de 2. Este dato concuerda con lo publicado en la bibliografía13,14. Debido a los niveles fluctuantes del factor de von Willebrand (VWF:Ag), la repetición de dichos niveles es obligatoria13. Hyatt et al., en su estudio de revisión de niños con evW1, realizan un mínimo de 2 analíticas. Reconocen las reservas a la hora de realizar más venopunciones confirmatorias, dada la corta edad de los pacientes y el estrés secundario14, porque no hay que olvidar que el estrés, el llanto y la ansiedad incrementan los niveles de VWF:Ag15. Pero aunque se intente limitar el número de venopunciones, parece difícil evitar un mínimo, ya que los propios criterios diagnósticos obligan a tener 2 determinaciones de laboratorio patológicas para su diagnóstico12.
En cuanto al tipo de sangrado, es llamativo que la mayoría de los niños estudiados no hayan presentado nunca sangrado (61,7%). Estudios actuales sobre el diagnóstico de coagulopatías recomiendan ser más restrictivos a la hora de iniciar un estudio de coagulopatía para evitar falsos positivos. Aconsejan adaptar y validar los cuestionarios sobre sangrado a la edad pediátrica. Este hecho es crítico para evitar estudios innecesarios en niños sanos16. Otro dato destacable es la presencia de sangrado en un 30,4% de los niños clasificados como sanos. Hay que recordar que la epistaxis y los hematomas son hallazgos frecuentes en niños sanos1.
Se han encontrado 3 niños diagnosticados de evW1 sin sangrado. Según la International Society on Thrombosis and Haemostasis (ISTH), estos casos serían clasificados como evW1 posible17. Como se comenta más adelante, los niños con evW1 pueden no haber sido sometidos a ningún desafío hemostático y ser esta la causa de no presentar sangrados patológicos14.
Un 33,3% de los niños con déficit de factor xii tienen sangrado. Nuestra interpretación es la misma que para los niños diagnosticados como sanos que presentan sangrados: son sangrados propios de la edad, sin relación con el déficit del factor xii. El único caso de hemofilia B tampoco presenta sangrados, probablemente porque el nivel del factor ix sea de un 45%.
En cuanto a la frecuencia de las coagulopatías diagnosticadas, como era esperable, la evW1 ha sido (junto con el déficit del factor xii) la coagulopatía más frecuente.
El diagnóstico de la evW1 continúa siendo un reto. Hay múltiples factores, como la edad, el grupo sanguíneo, las situaciones de estrés, los estados de inflamación y el propio embarazo, que contribuyen al cambio de los niveles plasmáticos del VWF:Ag. La clínica varía entre miembros afectos de una misma familia18. Además, los niveles plasmáticos del VWF:Ag usados como corte para el diagnóstico son una fuente de debate. Se han planteado diferentes cortes, desde el 15 al 50%, sin ningún consenso17,19. Usar el corte de 40% tanto para VWF:Ag como para el cofactor de la ristocetina parece evitar tanto el sobrediagnóstico como el infradiagnóstico20. Sin embargo, los actuales criterios de laboratorio de la ISTH proponen niveles de VWF:Ag inferiores al 20%17. Además del criterio del laboratorio, se requieren unos AF y una historia personal compatibles. El diagnóstico es definitivo si se cumplen los 3 criterios, y probable si falta alguno de los criterios de sangrado personal/familiar. Ya en 2000, Dean et al. publicaron los problemas de aplicar dichos criterios en pediatría: requería que los padres registrasen la duración exacta de todos los episodios hemorrágicos, la presencia de AF podía estar infravalorada al tener hermanos pequeños no sometidos a ningún desafío hemostático, y el propio paciente podía no mostrar un sangrado significativo por no haber presentado ningún desafío hemostático21.
También Hyatt et al. exponen que los criterios de la ISTH17 y del Hospital for Sick Children (HSC) son demasiado estrictos para los niños14. Dichos autores revisan retrospectivamente los casos diagnosticados de evW1 según los criterios del hematólogo en su hospital. Después aplican los criterios actuales de la ISTH y de la HSC: un 20% son diagnosticados de evW1 según los criterios de la ISTH y un 90% según los criterios de la HSC. Es importante aplicar criterios universales para el diagnóstico de la evW1. Y ese es un error que se comete en nuestra unidad. Pero deberían ser criterios adaptados a los niños.
El mayor número de pacientes diagnosticados de evW1 se ha obtenido dentro del grupo con TTPa alargado y clínica de sangrado, pero ha habido un caso dentro del grupo con TTPa y AP normales y con clínica de sangrado: la actividad del factor viii y, consecuentemente, el TTPa pueden tener valores normales cuando el VWF:Ag es mayor del 35%5.
En cuanto a la hemofilia A y B, en nuestra serie se ha diagnosticado un caso de hemofilia B, sin sangrado, con un factor ix>40%5,22.
Los trastornos de la coagulación raros representan un 3-5% del total de coagulopatías. El déficit del factor vii es el trastorno más frecuente dentro de este grupo, con una prevalencia de 1:500.00023. Sin embargo, en nuestra serie, de estos trastornos, el déficit más frecuente ha sido el del factor xi (2 casos). No se ha diagnosticado ningún déficit de factor vii. Posiblemente la alta prevalencia encontrada de déficit de factor xi se deba a que uno de esos pacientes, asintomático, se diagnosticó a raíz de un hermano diagnosticado previamente. En cuanto al déficit del factor x, el caso de nuestro estudio fue adquirido.
El riesgo de sangrado en pacientes con trastornos raros de la coagulación es menos predecible que en la hemofilia A y B24.
El déficit de precalicreína, HMWK y factor xii provoca in vitro un alargamiento del TTPa, que in vivo no se asocia a mayor riesgo de sangrado25. De hecho, algunos estudios evidencian que el déficit de factor xii incrementa el riesgo de trombosis venosa26. Su prevalencia en la población general es del 2,3% y podría ser la causa más común de TTPa prolongado inesperado durante las pruebas prequirúrgicas27. No existen datos en la población pediátrica, probablemente por su escasa repercusión clínica. En nuestra serie hemos encontrado 10 pacientes con este trastorno, 9 con déficit de factor xii y uno con déficit de otros factores de contacto, que representa una frecuencia del 21%. Su diagnóstico es relevante porque evita la realización de un elevado número de pruebas complementarias y la exposición innecesaria a hemoderivados.
LimitacionesSería necesario un estudio prospectivo y con un número mayor de pacientes para poder confirmar las conclusiones de forma satisfactoria.
Otra limitación se encuentra en el diagnóstico de la evW1. No se han usado los actuales criterios publicados por la ISTH o la HSC. Los niveles de Fc:VIII, el cofactor de la ristocetina y VWF:Ag considerados patológicos han sido del 45%, un límite superior a lo publicado en la actualidad. Además, no se ha tenido en cuenta el grupo sanguíneo para ajustar los niveles del VWF:Ag. Probablemente estas circunstancias hayan producido un sobrediagnóstico de la evW1.
ConclusionesEl motivo más frecuente para iniciar un estudio de coagulopatía ha sido el hallazgo de un TTPa alargado sin clínica de hemorragia. Probablemente se tenga que ser más restrictivo desde los servicios que remiten pacientes para estudio. Ayudaría utilizar cuestionarios de sangrado adaptados a la infancia y una buena exploración física e historia personal/familiar en busca de signos que indiquen sangrados patológicos.
Se deben conocer las peculiaridades de los patrones de sangrado fisiológicos en los niños para evitar falsos diagnósticos de coagulopatías.
Aunque no ha sido el tema de este artículo, creemos que la evW no tiene aún unos criterios universales aceptados para la infancia.
Y, por último, se debe relacionar siempre la clínica con el laboratorio: un TTPa y una AP normales con sagrados patológicos obligan a ampliar el estudio.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo se ha presentado previamente en el Congreso de la AEP de Sevilla, 2013.
