





Symptoms/signs suggestive of coagulopathy is a frequent complaint in Paediatric Haematology units. Both the clinical and family history are essential for diagnosis.
Patients and methodsRetrospective and descriptive study of patients referred to a Paediatric Haematology unit of a tertiary hospital for possible coagulopathy during 2012.
ResultsA total of 47 children were studied, of whom 61.7% had not previously presented bleeding. The most frequent reason for referral was a prolonged activated partial thromboplastin time without any haemorrhage (42.5%), of these, 25% were diagnosed with coagulopathy with a real risk of bleeding. Patients referred due to a prolonged activated partial thromboplastin time with bleeding more frequently (41.7%) present coagulopathy with a real risk of bleeding. Children with a family history of bleeding are diagnosed more frequently with coagulopathy with real risk of bleeding: 37.5% (family history) vs. 14.3% (no family history). The most frequent diagnoses were: healthy children (48.9%), von Willebrand type 1 disease (19.1%), factor xii deficiency (19.1%), factor xi deficiency (4.2%), prekalikrein/high molecular weight kininogen deficiency (2.1%), acquired deficiency of factor x (2.1%), and factor ix deficiency (2.1%).
ConclusionsA thorough personal and family bleeding history and physical examination are the first steps for a correct differential diagnosis. The reason for referral should be based more on clinical bleeding and not just on an abnormal coagulation time. The most frequent diagnoses were type 1 von Willebrand disease and factor xii deficiency.
Los síntomas/signos indicativos de una coagulopatía son un motivo de consulta frecuente en las unidades de Hematología Pediátrica. Tanto la clínica como los antecedentes familiares son fundamentales para el diagnóstico.
Pacientes y métodosEstudio retrospectivo y descriptivo de los pacientes derivados a una consulta de Hematología Pediátrica de un hospital de tercer nivel por posible coagulopatía durante el año 2012.
ResultadosSe estudiaron 47 niños. El 61,7% no había presentado previamente sangrado. El motivo de derivación más frecuente fue un tiempo de tromboplastina parcial activada alargado sin hemorragia (42,5%); de estos, un 25% fue diagnosticado de una coagulopatía con riesgo real de sangrado. En los pacientes derivados por tiempo de tromboplastina parcial activada alargado con clínica hemorrágica se detecta una coagulopatía con riesgo real de sangrado con mayor frecuencia (41,7%). En los niños con antecedentes familiares de sangrado se diagnostica con más frecuencia una coagulopatía con riesgo real de sangrado: 37,5 vs. 14,3% (niños sin antecedentes familiares). Los diagnósticos han sido: sano (48,9%), enfermedad de von Willebrand tipo1 (19,1%), déficit de factor xii (19,1%), déficit de factor xi (4,2%), déficit de precalicreína/cininógeno de alto peso molecular (2,1%), déficit adquirido de factor x (2,1%) y déficit de factor ix (2,1%).
ConclusionesLos antecedentes personales y familiares de sangrado orientan el diagnóstico de una coagulopatía. El motivo de derivación debería basarse en mayor medida en la clínica hemorrágica y no solo en un tiempo de laboratorio alterado. Los diagnósticos más frecuentes han sido enfermedad de von Willebrand tipo 1 y déficit de factor xii.
Coagulopathy-indicative signs are a frequent reason for consultation in haematology. Its diagnosis can be difficult, due to specific bleeding age patterns: epistaxis (39%) and easy bruising (24%) are common without any haemorrhagic disorder. However, these can be the only clinical symptoms in children with coagulopathies who have not undergone any haemostatic challenge.1
In addition, differential bleeding/bruising diagnosis in children includes other entities, such as platelet alterations, vasculopathies and abuse.
Among hereditary coagulopathies, the most frequent is von Willebrand disease (vWD), which has a prevalence of approximately 1%, according to studies.2,3 More recent studies, however, have reported a 0.1% prevalence of clinically symptomatic vWD in children.4 This is followed by factor viii (haemophilia A) and factor ix (haemophilia B) deficiency, with a predominance of 1:5000 males and 1:30,000 males, respectively.5
Some factor deficiencies do not result in bleeding, such as the deficiency of some contact factors: high molecular weight kininogen (HMWK), prekallikrein and factor xii. These factors are not involved in the propagation phase of the coagulation cascade and, therefore, their deficiency, in spite of prolonging the activated partial thromboplastin time (APTT) in vitro, is not associated with bleeding in vivo.6
In this study, we set out to determine which services refer the greatest number of patients for coagulation analysis and the reason for referral, to correlate laboratory findings with clinical symptoms of bleeding, and to determine the most common diagnoses made in our hospital. Greater insight into these patients will help improve diagnosis protocols.
Patients and methodsRetrospective descriptive analysis of children under study due to suspected coagulopathy at the paediatric oncohaematology unit of the Virgen Macarena University Hospital (UH) in Seville in 2012.
At this unit, in 2012, all paediatric solid/haematological tumours (except for brain tumours) and non-oncologic haematology disorders were studied and treated. Patients diagnosed with haemophilia A or B were referred to the Haemophilia Unit from the Rocio UH in Seville after diagnosis.
There were 20 new oncology patients in 2012, and 120 haematological patients.
Clinical treatment was given by paediatric haematologists and laboratory techniques by haematologists.
We have taken into consideration children with altered coagulation times, those with normal coagulation and bleeding times (after ruling out thrombocytopenia), and those studied due to having a family member with a hereditary coagulopathy. A total of 52 children were studied. Of these, 5 were oncology patients undergoing active treatment and were excluded due to the potential interactions between chemotherapy and the underlying disease.
The other 47 children were ultimately selected.
The diagnosis of coagulopathy was based on a factor plasma level of less than 45%, and in the case of vWD1, on a ristocetin co-factor of less than 45%, and a relevant clinical/family history (FH).
Factor xii, prekallikrein and HMWK deficiencies were considered as having no real risk of bleeding.
The following variables were collected: age, gender, service making the referral, reason for the referral, FH of coagulopathy/bleeding, presence and origin of previous clinical symptoms of bleeding, number of blood tests performed and final diagnosis.
A positive family history was taken into consideration if there was significant cutaneous and/or mucous bleeding in a first-degree family member. No standardised bleeding questionnaires were used.
All the data that were obtained and considered as study variables were stored and analysed with the statistical software package SPSS® v.15.0 (SPSS Inc., Chicago, IL, USA).
Descriptive statistical analysis: qualitative variables are expressed as absolute frequencies and percentages. Quantitative variables as mean, median and range.
Analytical statistical analysis: Fisher's exact test was used to compare the frequencies of 2 qualitative variables, the chi-square goodness of fit test was used to compare frequencies of the same qualitative variable, and Pearson's Chi-square test was used to analyse the association between 2 qualitative variables. A value of p<0.05 was considered significant.
ResultsData from 47 children were collected: 28 boys (59.6%) and 19 girls (40.4%).
The mean age at the first visit was 6.1 years (median: 6 years; interval: 1 month-13 years).
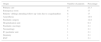
Patients’ origin is shown in Table 1.
Origin of the children under study due to coagulopathies.
| Origin | Number of patients | Percentage |
|---|---|---|
| Primary care | 10 | 21.3 |
| Emergency room | 8 | 17 |
| Patients’ siblings attending follow-up visits due to coagulopathies | 8 | 17 |
| Anaesthesia | 7 | 14.9 |
| Paediatric surgery | 6 | 12.8 |
| Hospitalisation unit | 3 | 6.4 |
| Paediatric oncology | 1 | 2.1 |
| Neonatology | 1 | 2.1 |
| IC paediatric unit | 1 | 2.1 |
| Dentistry | 1 | 2.1 |
| ENT | 1 | 2.1 |
A confirmatory diagnosis was based on the mean of 2 blood tests (median: 2; interval: 1–5).
Haemorrhagic clinical symptoms and diagnoses:
- •
Eighteen of the 47 patients (38.3%) had a history of bleeding, mainly epistaxis (12 children, 66.6%).
- •
No diagnosis was possible in 1 study patient who did not return for subsequent appointments. Final diagnoses, presence/absence of bleeding and statistically significant differences between bleeding/no bleeding are summarised in Table 2.
Table 2.Final diagnoses and existence/absence of haemorrhagic clinical symptoms.
Diagnosis Bleeding Number of patients Percentage pa Healthy 23 48.9 Yes 7 30.4 0.061 No 16 69.6 VWD1 9 19.1 Yes 6 66.7 0.058 No 3 33.3 Factor XII deficiency 9 19.1 Yes 3 33.3 0.317 No 6 66.7 Factor xi deficiency No 2 4.2 Contact factor deficiency (other than xii) No 1 2.1 Acquired factor x deficiency Yes 1 2.1 Factor ix deficiency No 1 2.1 Lost Yes 1 2.1
FH of bleeding:
- •
Two patients were adopted and these data could not be gathered.
- •
Table 3 summarises the presence/absence of an FH of bleeding according to final diagnosis. FH of bleeding is more uncommon in children diagnosed with coagulopathy with no real risk of bleeding (p=0.029).
Table 3.Family history of bleeding.
Final diagnosis Total (n=45) With an FH of bleeding (n=24) No FH of bleeding (n=21) p* Healthy 23 (51.1) 13 (56.5) 10 (43.5) NSa Coagulopathy with no risk of bleeding 10 (22.1) 2 (20) 8 (80) 0.029a Coagulopathy with risk of bleeding 12 (26.7) 9 (75) 3 (25) 0.07a FH, family history; NS, non-statistically significant differences.
Data expressed as n (%).
Reason for referral:
- •
This is summarised in Table 4.
Table 4.Reason for the referral.
Reason for the referral Number of patients Percentage Prolonged APTT 33 70.2 No bleeding 20 66.6 Bleeding 13 39.4 Patients’ siblings attending follow-up visits due to coagulopathies 10 21.3 No bleeding 9 90 Bleeding 1 10 Bleeding with APTT and PA normal levels Bleeding 2 4.3 Bleeding with reduced PA Bleeding 2 4.3 PA, prothrombin activity; APTT, activated partial thromboplastin time.
Analysis per group is as follows:
- -
Prolonged APTT with no bleeding: 20 children. The final diagnosis was: 8 (40%) healthy children, 6 (30%) with factor xii deficiency (one of whom was diagnosed with lupus anticoagulant within a context of sepsis), 3 (15%) with vWD1, 1 (5%) with factor xi deficiency, another (5%) with deficiency of other contact factors (HMWK, prekallikrein) and lastly, 1 (5%) with factor ix deficiency.
- -
Prolonged APTT with bleeding: 13 children, although 1 was lost to follow-up. Below is a summary of the type of bleeding and of the final diagnosis:
- (a)
Epistaxis: 10 (77%). The final diagnosis was: 5 patients (50%) with vWD1 (1 of whom was diagnosed with lupus anticoagulant); 2 patients (20%) with factor xii deficiency; 2 (20%) were healthy.
- (b)
Haemorrhage at another level: 3 (23.1%). The final diagnosis was: 2 healthy children (66.7%), who had been referred due to haematemesis in 1 case and petechiae in the other case; and 1 patient with factor xii deficiency (33.3%). This patient had presented intraventricular haemorrhage within a prematurity context.
- (a)
Excluding 1 patient that was lost to follow-up and 1 premature infant, 5 (45.5%) of the 11 patients referred due to an altered APTT and bleeding were diagnosed with coagulopathy with a high risk of bleeding. In the group of children with a prolonged APTT with no clinical symptoms of bleeding, 5 out of 20 (25%) were diagnosed with coagulopathy with a high risk of bleeding. These differences, however, are not statistically significant.
- -
Hereditary haemorrhagic disorder: 10 children. The final diagnoses were:
- (a)
Healthy: 9 (90%). Eight with no bleeding and one with occasional epistaxis.
- (b)
Factor XI deficiency: 1 patient (10%) with no bleeding.
- (a)
- -
Bleeding with normal APTT and prothrombin activity (PA): 1 child was diagnosed with vWD1 after presenting capillary haemorrhage following a phimosis intervention. Another child, who was referred due to epistaxis and haemorrhage after a dental extraction, was not diagnosed with any alteration.
- -
Bleeding with reduced PA: 1 child with prolonged APTT with clinical symptoms of rectal bleeding was diagnosed with factor x deficiency caused by vitamin K deficiency. In another child with recurrent haematomas, no pathology was diagnosed.
The results of the study showed that, in our Paediatric Haematology Unit, most of the children who were referred to rule out coagulopathy were referred due to prolonged APTT with no clinical haemorrhagic symptoms, and that 75% of these and 48.7% of the total of children under study were healthy.
Was the referral justified? Is it necessary to perform coagulation studies in patients with no bleeding history? Our findings show room for improvement in the protocols for referring patients to the Haematology Unit. When should basic coagulation analysis be performed? And which patients with altered times should have further analysis? Referrals from Surgery and Anaesthetics Units, together, represent the most frequent reason for consultation: APTT and PA studies are a part of the preoperative work-up without taking into consideration the patient's personal history or FH of bleeding.
The American Academy of Otolaryngology-Head and Neck Surgery recommends preoperative coagulation test only if a coagulopathy is suspected based on the patient's personal or family history.7 Likewise, the British Committee for Standards in Haematology recommended in 2008 obtaining a complete bleeding history instead of performing universal coagulation screening.8 Cooper et al. found not performing coagulation tests before amygdalectomy in children to be the most cost-effective strategy.9 After amygdalectomy, one of the most frequent interventions in children, Zagólski detected bleeding in several patients with previously normal coagulation tests, and none in patients with a previously altered APTT. These authors emphasise that the incidence of haemorrhage after an amygdalectomy does not increase in patients with a prolonged APTT, and a personal history of recurring epistaxis; haematomas after minor injury and prolonged haemorrhage after minor injuries are better predictors of postoperative bleeding.10 Hubner concluded that a detailed and supervised history has great predictive value in the diagnosis of coagulopathies. The absence of bleeding predicts that laboratory results will be normal in about 94% of the cases.11
Nevertheless, how would coagulopathies with no haemorrhage symptoms be diagnosed if no laboratory tests are performed? According to Bowman et al., the absence of bleeding in a child with vWD can be due to the lack of surgical interventions, dental extractions or menstruation that can provoke haemorrhage.4 In these cases, FH plays a crucial role in diagnosis. Furthermore, the diagnosis of certain coagulopathies, such as vWD, requires the presence of a positive FH.12
Our study design is too limited to allow us to draw conclusions on FH of bleeding. We can only state that an FH of bleeding was more common in the group of children diagnosed with coagulopathies and without a high risk of bleeding.
On average, 2 blood tests were performed per patient in our study, a rate consistent with other studies.13,14 Due to the fluctuating levels of von Willebrand factor (VWF:Ag), a repeat test is mandatory.13 Hyatt et al., in their revised study on children with vWD1, performed a minimum of 2 blood tests. Particular care is needed when drawing blood for follow-up tests in paediatric patients, due to the risk of secondary stress14; it should be borne in mind that stress, crying and anxiety increase vWF:Ag levels.15 Nevertheless, 2 separate blood tests are needed to confirm diagnosis of vWD, so at least 2 venipunctures must be performed.12
As for the type of bleeding, it is interesting to note that most children under study never presented with bleeding (61.7%). Current studies on the diagnosis of coagulopathies recommend a more restrictive approach to coagulopathy analyses to avoid false positives. They recommend adjusting and validating questionnaires on paediatric bleeding. This is crucial to avoid unnecessary studies in healthy children.16 Another remarkable finding is the existence of bleeding in 30.4% of the children classified as healthy. In this regard, it should be borne in mind that epistaxis and haematomas are common findings in healthy children.1
Three children diagnosed with vWD1 without bleeding were found. According to the International Society on Thrombosis and Haemostasis(ISTH), these cases would be classified as potential vWD1.17 As mentioned above, children with vWD1 might not have been subjected to a haemostatic challenge, and this could explain why they did not present with pathological bleeding.14
In our study, 33.3% of children with factor xii deficiency presented bleeding. Our interpretation is the same as in children with bleeding who were diagnosed as healthy: bleeding is frequent in children, regardless of a factor xii deficiency. The only case of haemophilia B did not exhibit bleeding, probably because the level of factor ix was 45%.
Regarding the frequency of coagulopathies, as expected, vWD1 (together with factor xii deficiencies) was the most frequent.
The diagnosis of vWD1 is still a challenge. Multiple factors, such as age, blood group, stress situations, inflammatory conditions and pregnancy itself, contribute to alterations in VWF:Ag plasma levels. Clinical symptoms vary among affected members of the same family.18 In addition, VWF:Ag plasma levels used as a cut point for diagnosis are controversial. Several cut points have been proposed, from 15% to 50%, with no consensus.17,19 Using a 40% cut point for both VWF:Ag and ristocetin co-factor seems to prevent both over- and under-diagnoses.20 However, the current laboratory criterion proposed by the ISTH is VWF:Ag levels below 20%.17 In addition to laboratory criteria, there must be a compatible FH and personal history. The diagnosis is definite if 3 criteria are fulfilled, and is only suggestive if some of the bleeding criteria, whether individual or family, are not fulfilled. In 2000, Dean et al. published a study comparing the application of these criteria in paediatrics: parents must register the exact duration of all haemorrhagic episodes; the existence of an FH could be underrated if the patient has younger siblings who have not undergone any haemostatic challenge; and the patient might not have exhibited significant bleeding due not having undergone haemostatic challenges.21
Hyatt et al. also stated that the ISTH17 and Hospital for Sick Children (HSC) criteria are too strict for children.14 These authors retrospectively reviewed cases diagnosed with vWD1 according to the criteria of the hospital's haematologist. Afterwards, they applied the current ISTH and HSC criteria: 20% were diagnosed with vWD1 according to the ISTH criteria, and 90% according to the HSC criteria. It is important to apply universal criteria to diagnose vWD1, and this has not been done in our unit. But even more importantly, the criteria must be adapted to children.
The greatest number of patients diagnosed with vWD1 were from the group with a prolonged APTT and clinical symptoms of bleeding, but there was a case from the group with normal APTT and PA with clinical symptoms: factor viii activity, and, consequently, APTT levels can be normal when VWF:Ag is higher than 35%.5
As for haemophilia A and B, in our series we diagnosed 1 haemophilia B case, without bleeding, with a factor ix >40%.5,22
Rare coagulation disorders represent between 3% and 5% of all coagulopathies. Factor vii deficiency is the most frequent disorder within this group, with a predominance of 1:500,000.23 However, in our series, the most common deficiency was factor xi deficiency (2 cases); no factor VII deficiencies were found. The high prevalence of factor XI deficiency could be due to the fact that one such patient, who was asymptomatic, was diagnosed following a diagnosis of this deficiency in a sibling. The factor X deficiency patient in our study was an acquired case.
The risk of bleeding in patients with rare coagulation disorders is less predictable than in haemophilia A and B.24
Prekallikrein, HMWK and factor xii deficiencies cause in vitro APTT prolongation, which is not associated with a higher risk of bleeding in vivo.25 In fact, some studies show that a factor xii deficiency increases the risk of venous thrombosis.26 Its prevalence in the general population is 2.3% and it could be the most frequent cause of an unexpected prolonged APTT during preoperative workup.27 There are no data in the paediatric population, probably due to its limited clinical manifestation. In our series, we found 10 patients with this disorder, 9 with factor xii deficiency and 1 with a deficiency of other contact factors, which represents a frequency of 21%. Diagnosis of these disorders will avoid complementary tests and unnecessary exposure to blood products.
LimitationsIt would be necessary to perform a prospective study with a higher number of patients to be able to confirm these conclusions satisfactorily.
Another limitation lies in the diagnosis of vWD1. We did not use the current criteria published by ISTH or HSC. Fc:VIII, ristocetin co-factor and VWF:Ag of 45% were considered pathological. This threshold is higher than that recommended in the literature. In addition, we did not take into consideration the blood group to adjust the levels of VWF:Ag. It is possible that these circumstances have generated an over-diagnosis of vWD1.
ConclusionsThe most frequent reason to initiate a coagulopathy analysis has been the existence of a prolonged APTT with no clinical symptoms of haemorrhage. More restrictive protocols may be needed in services referring patients for coagulopathy studies. It would be helpful to use questionnaires on bleeding adapted to children and perform thorough physical examinations and personal/family history, looking for signs that point to pathological bleedings.
It is important to be familiar with bleeding patterns in children in order to avoid false coagulopathy diagnoses.
Although the topic is beyond the scope of this study, we believe that no universally accepted criteria for the diagnosis of vWD have yet been established.
Finally, clinical symptoms must always correlate with laboratory results: normal levels of APTT and PA with pathological bleeding require further analyses.
Conflict of interestsThe authors declare that there are no conflicts of interest.
Please cite this article as: Romero I, Conde N, Aldana DG, Ruano A, Fernández-Teijeiro A. Revisión de los pacientes estudiados por coagulopatía en una unidad de Oncohematología. An Pediatr (Barc). 2016;84:85–91.
