


Los angiomas cavernosos son espacios vasculares sinusoidales, separados por finas paredes de colágeno, sin parénquima cerebral interpuesto. Tienen naturaleza dinámica, siendo la hemorragia el factor fundamental para su crecimiento. Su riesgo funcional y/o vital depende de su localización y evolución1,2. Localizados el 75-80% supratentoriales y el 18-35% en troncoencéfalo.
Se clasifican según el número de lesiones (únicos/múltiples) y su patrón hereditario (esporádicos/familiares)2. Afectan al 0,4-0,8% de la población1, y representan el 10-20% de las lesiones vasculares cerebrales3.
La cavernomatosis múltiple (CM) (OMIM#116860) se caracteriza por la presencia de múltiples cavernomas localizados en el sistema nervioso central (SNC). La forma familiar presenta herencia autosómica dominante, con penetrancia y expresividad clínico-radiológica variables. Hasta la fecha hay 3 genes descritos en su génesis: CCM1/KRIT1 (7q21-22), CCM2/malcaverina (7p13) y CCM3/PDCD10 (3q26.1) con penetrancias del 88, 100 y 63%, respectivamente4.
El número de cavernomas incrementa con la edad y en los casos familiares5, especialmente en mutaciones en CCM1. Los casos familiares por CCM3 presentan menos individuos afectos, pero mayor riesgo de sangrado y comienzo más precoz6.
ClínicaGeneralmente son asintomáticos (15-20%). Los casos familiares son asintomáticos hasta el 40%. Suelen manifestarse entre la 2.ª y 5.ª décadas de la vida. Sin diferencia entre sexos, con presentación hemorrágica en niños y silentes en ancianos1.
Existen 3 patrones de presentación:
- –
Crisis epilépticas
- –
Déficit neurológico focal: déficit de pares (69%), alteraciones de la sensibilidad (39%), déficit motor (38%) o ataxia (30%)
- –
Cefalea
Los factores de riesgo asociados a comportamiento más agresivo son: gestación, sexo femenino, casos múltiples o familiares, cavernomas de tronco, exéresis parciales y tras radioterapia1.
Su comportamiento suele ser benigno. Lo que debe hacer plantear una actitud conservadora, especialmente si son múltiples2.
DiagnósticoEl 50% es hallazgo casual por neuroimagen2. Siendo la RM la técnica de elección.
Ante pacientes con malformaciones vasculares en SNC o retina, debe plantearse la posibilidad de una CM. Incluso evaluando a familiares de primer grado, si el índice presentase múltiples malformaciones o hubiera historia familiar sugestiva (epilepsia, malformaciones vasculares cutáneas, sangrados intracraneales o muertes súbitas inexplicadas). Un diagnóstico presintomático permitiría un seguimiento más estrecho, y plantear un tratamiento lo más precoz posible si fuera necesario7.
TratamientoQuirúrgico: indicado de forma individualizada. Considerándolo en cavernomas sintomáticos, siempre que sean accesibles y puedan extirparse completamente. Exéresis parciales presentan riesgo superior de resangrado y peores resultados que el tratamiento no quirúrgico2,8.
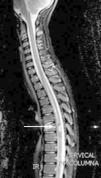
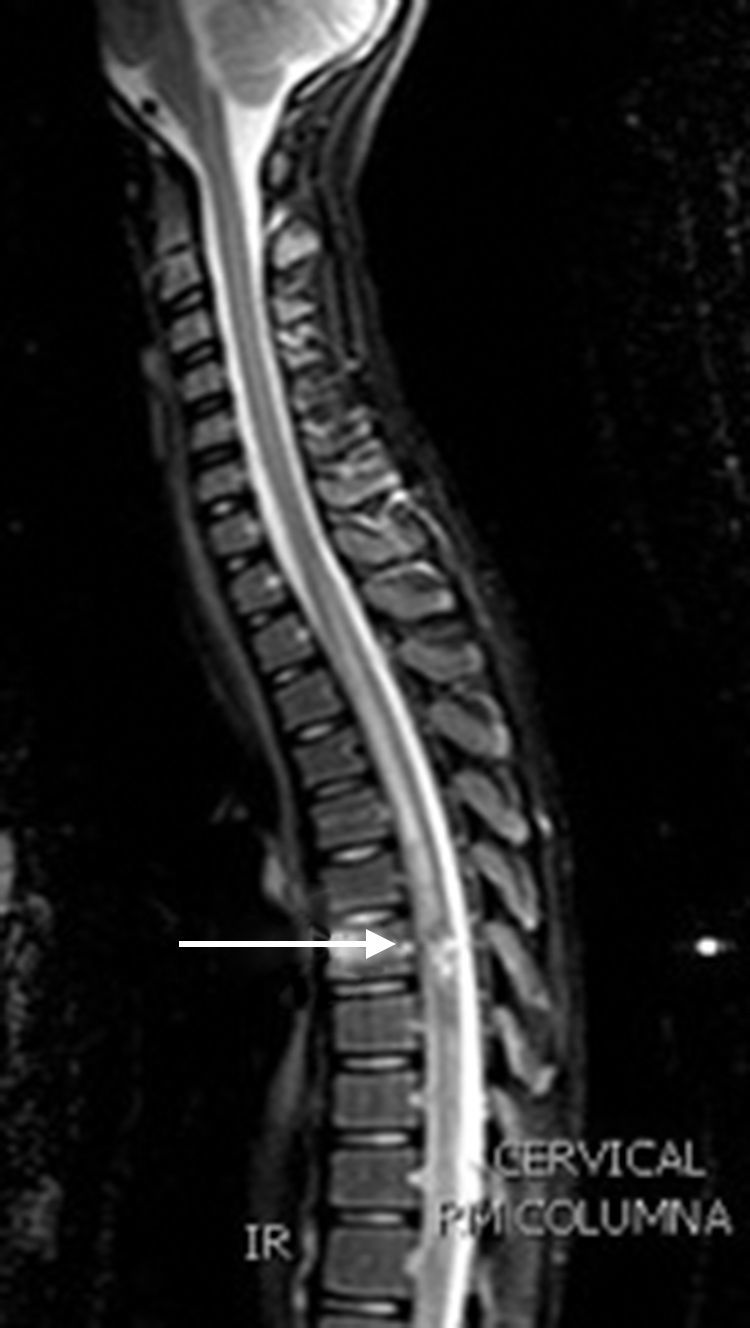
Caso clínicoNiño de 10 años, sin antecedentes y normal desarrollo psicomotor. Presenta empeoramiento del estreñimiento habitual, dolor abdominal e imposibilidad para la micción en las últimas 24h; con anestesia del pie derecho, aparición progresiva de parestesias en extremidad inferior derecha (EID) y dificultad progresiva para la deambulación. Exploración: debilidad y disminución del tono en EID. Hemograma y bioquímica sanguíneas con LDH, las radiografías abdominal y de columna lumbar, y la TAC abdómino-pélvica fueron normales. Con sospecha de mielopatía compresiva, y ante progresión de la debilidad en EID, con imposibilidad para la deambulación, se realiza RM medular, donde se aprecia lesión tumoral en T7 (fig. 1). Se inicia tratamiento con dexametasona intravenosa a dosis altas, progresando la debilidad a la otra extremidad, y se decide realizar cirugía descompresiva medular; previo a esta, la RM cráneo-medular muestra múltiples lesiones compatibles con cavernomas (fig. 2).
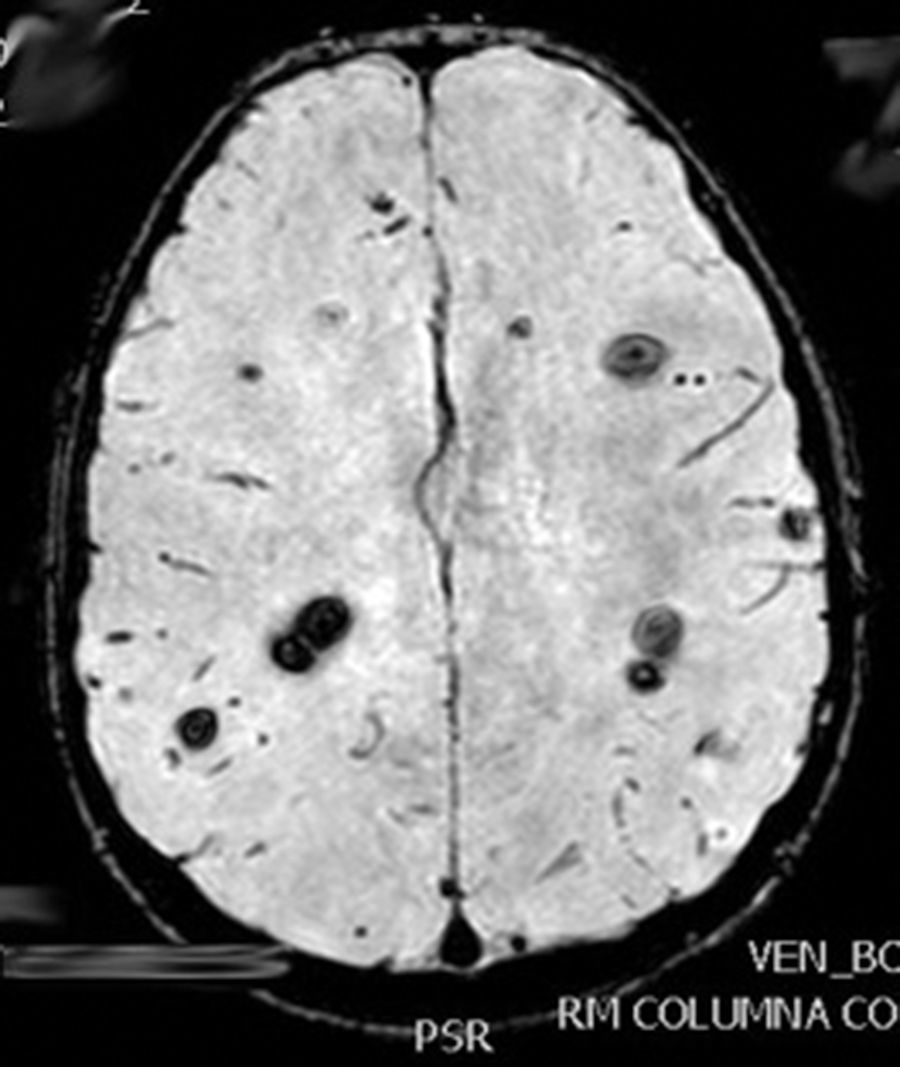
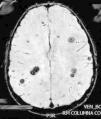
La lesión medular es resecada completamente, y su análisis confirma un cavernoma. Reanamnesiando, se nos refiere que en una TAC realizada a la madre 22 años antes, se le dijo que presentaba múltiples lesiones, sin poder precisar su naturaleza, permaneciendo asintomática.
La evolución fue satisfactoria, con recuperación progresiva de la función vesical, fuerza en EID y la deambulación autónoma. La RM cerebral materna confirmó la CM. El estudio genético en nuestro paciente, demostró una mutación en CCM1/KRIT1, en heterocigosis, c.418 C>T, que produce un cambio en la pauta de lectura que da lugar a la aparición de un codón STOP (p.Arg140*). No descrito previamente.
ConclusionesLa compresión medular es una urgencia neurológica que obliga a la realización inmediata de estudios, y a la toma de decisiones terapéuticas que pueden marcar el pronóstico neurológico.
La CM es un diagnóstico infrecuente en la edad pediátrica, puede permanecer asintomático o producir clínica según localización de las lesiones. Los antecedentes familiares son importantes para la sospecha clínica y el diagnóstico.
Un correcto diagnóstico permite ofrecer un adecuado consejo genético. Un diagnóstico presintomático permitiría un seguimiento más estrecho y plantear un tratamiento lo más precoz posible si fuera necesario7.
En su tratamiento, la actitud conservadora mediante observación clínica y neuroimagen periódicas es la más importante en los pacientes asintomáticos. Reservando la cirugía ante lesiones sintomáticas, accesibles y que puedan ser resecadas completamente2,8.
Caso clínico presentado como comunicación tipo póster en el 62° Congreso Nacional de la AEP, Sevilla, 6-8 junio de 2013.
