


El síndrome de resistencia a hormonas tiroideas (RTH) es una entidad clínica poco frecuente en la que el trastorno por déficit de atención con hiperactividad (TDAH) puede ser la única o principal manifestación1.
Presentamos el caso de un niño con retraso ponderal que asoció en su evolución un TDAH. Antecedentes personales: parto a las 37 semanas, PRN 2.980g, LRN 49cm, PC 32,5cm (cm (−1,5DE), peso 10,9kg (-2,0DE), IMC14,0kg/m2 (−1,7DE), tensión arterial (TA) y frecuencia cardiaca (FC) normales. En el estudio realizado se observó una elevación de los niveles de T4 libre de 3,2ng/dl (0,8-2), con niveles normales de TSH. Estos resultados fueron confirmados, siendo los niveles de T3 de 351ng/dl (80-160). El valor de la tiroglobulina, los anticuerpos anti-TPO, antitiroglobulina y antirreceptor de TSH fueron negativos (tabla 1). La ecografía cervical mostró una glándula tiroidea de tamaño y ecoestructura normales.
Seguimiento de la función tiroidea en el paciente con síndrome de resistencia a hormonas tiroideas
| Dos años y 6 meses | Dos años y 8 meses | Tres años y 6 meses | Cuatro años y 7 meses | Seis años | |
| T4 (ng/dl) | 3,2 (VN 0,8-2) | 3,4 (VN 0,8-2) | 2,1 (VN 0,6-1,4) | 2,0 (VN 0,6-1,4) | 2,1 (VN 0,6-1,4) |
| T3 (ng/dl) | 351 (VN 80-160) | 229 (VN 80-160) | 233 (VN 80-160) | ||
| TSH ((U/ml) | 2,59 (VN 0,5-4,5) | 3,0 (VN 0,5-4,5) | 1,28 (VN 0,5-4,5) | 1,59 (VN 0,5-4,5) | 1,3 (VN 0,5-4,5) |
| TG (ng/ml) | 78,1 (VN 0,1-35) | ||||
| Anti-TPO (UI/ml) | VN | VN | VN | VN | |
| Anti-TG (UI/ml) | VN | VN | VN | VN | |
| Antirreceptor de TSH (UI/l) | VN | VN | VN |
Anti-TG: anticuerpos antitiroglobulina; Anti-TPO: anticuerpos antiperoxidasa; TG: tiroglobulina; VN: valores normales.
Los desarrollos físico y psicomotor del paciente han sido normales. No ha manifestado síntomas de hipertiroidismo ni bocio. Los niveles de hormonas tiroideas han persistido aumentados, y los de TSH, en rango normal (tabla 1). A los 5 años presentó un defecto de concentración en el colegio con retraso en la lectoescritura y un año después fue diagnosticado de TDAH, presentando buena respuesta al tratamiento con metilfenidato (5mg cada 12h). Su cociente intelectual es normal. La RM craneal no muestra alteraciones hipofisarias o hipotalámicas. En la revisión realizada a los 6 años y 3 meses, su talla es de 115,4cm (−0,6DE), peso 17,4kg (−1,5DE), IMC 13,1% (−1,5DE), PC 49,5cm (−2,0DE), TA, FC y exploración general normales.
Antecedentes familiares: madre con bocio con nódulos tiroideos y niveles de T4 libre 1,6ng/dl (0,6-1,4), T3 165ng/dl y TSH0,50mUI/l (0,5-4,5), PC de −1,9DE y antecedentes de mal rendimiento escolar. La abuela, 2 tías maternas y el hermano presentan niveles elevados de hormonas tiroideas. El padre no presenta patología tiroidea.
El estudio genético-molecular del gen del receptor beta de hormonas tiroideas (THRβ) en nuestro paciente confirma la presencia de una mutación heterocigota M442V en el exón 10 diagnóstica del RTH (fig. 1). Esta mutación, previamente descrita, se sabe que produce alteraciones en la funcionalidad del receptor.
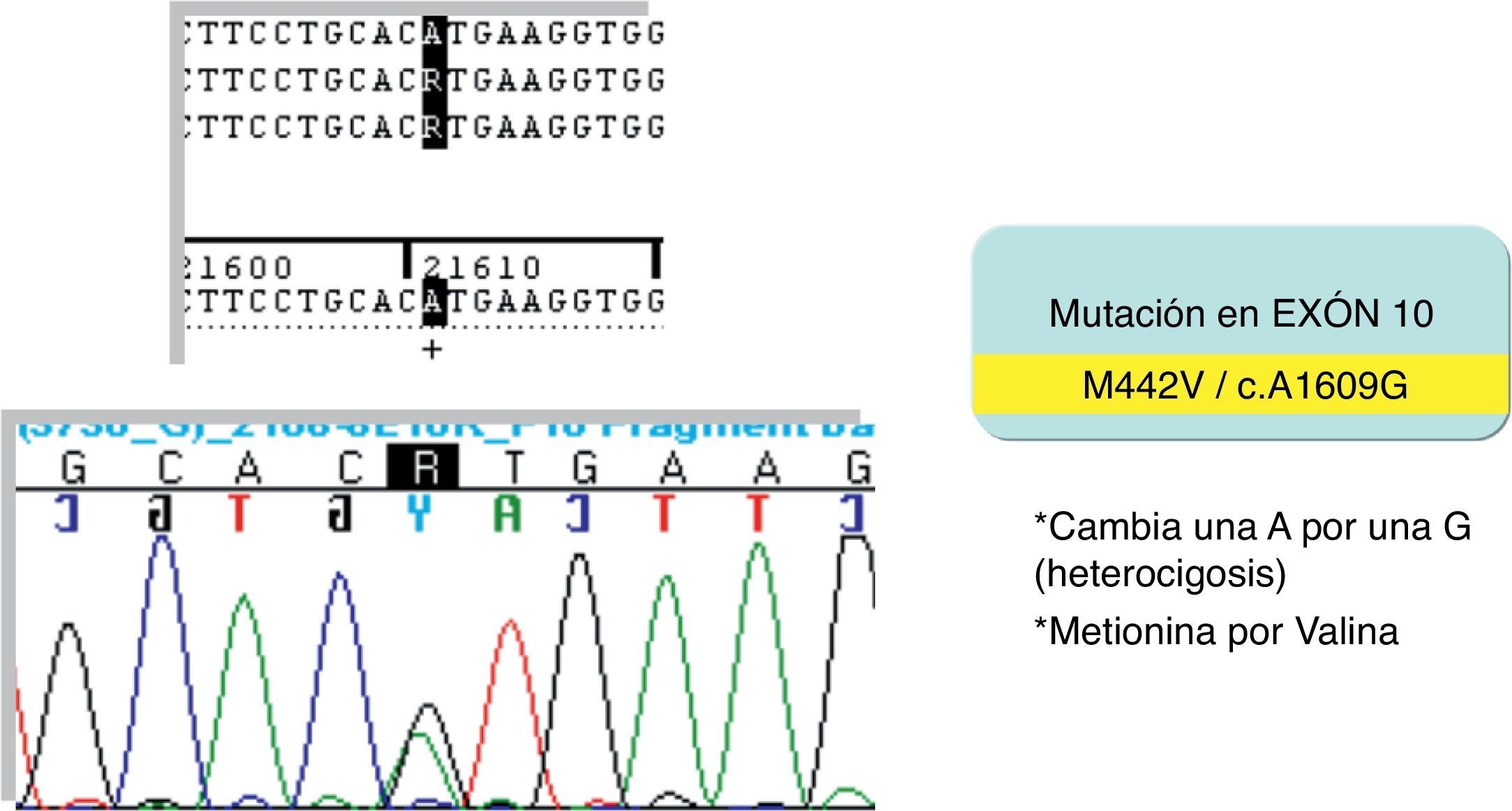
El RTH es un síndrome hereditario caracterizado por la disminución de la respuesta de los órganos diana a las hormonas tiroideas. Fue identificado por primera vez en 19672 y es secundario a mutaciones en el gen THRβ. Sigue una herencia autosómica dominante con la excepción del caso original descrito por Refetoff3,4.
La incidencia del RTH es baja: 1:40.000-50.000 nacidos vivos. La expresión es variable, estando los pacientes eutiroideos o presentando síntomas que van desde el hipotiroidismo al hipertiroidismo según el grado de resistencia en los tejidos. El hallazgo clínico más frecuente es el bocio (65-95%). Se describe taquicardia en un 33-75%, trastornos del aprendizaje con retraso escolar, alteraciones del lenguaje y TDAH en un 40-60%, y a veces talla baja, retraso de dentición e hipoacusia1. El síndrome está asociado con un riesgo aumentado de enfermedad tiroidea autoinmune5.
Se debe sospechar en pacientes que presentan niveles séricos elevados de T3 y T4 con valores de TSH normales o elevados. La confirmación diagnóstica se realiza mediante el estudio genético-molecular del THRβ3,4, localizado en el cromosoma 3. En un 85% de los casos de RTH se demuestran mutaciones en el gen THRβ (descritas más de 100 diferentes). En el 15% de casos familiares restantes no se encuentran mutaciones y se piensa que la RTH es secundaria a mutaciones en los genes que codifican los cofactores que interactúan con el receptor6,7.
Los pacientes con RTH y eutiroidismo compensado no precisan tratamiento3,4. Aquellos con hipotiroidismo precisan tratamiento sustitutivo con levotiroxina, y los que presentan síntomas de hipertiroidismo, como taquicardia o temblor, responden a la administración de betabloqueantes. El TDAH debe tratarse con los fármacos disponibles para esta indicación, aunque se han comunicado casos aislados de tratamiento con liotironina (L-T3) en dosis suprafisiológicas para reducir la hiperactividad e impulsividad cuando la respuesta es insuficiente, o con el derivado acético de la T3 (TRIAC)8,9.
En conclusión, el TDAH puede ser la principal manifestación clínica de un RTH. Aunque esta es una entidad poco frecuente, debemos sospecharla ante una elevación persistente de hormonas tiroideas con niveles de TSH normales o elevados. El tratamiento sería el del TDAH.
