


Caso clínico
Niña de 23 meses que acude derivada por su pediatra por pubarquia de 3 meses de evolución. La madre refiere, coincidiendo con la pubarquia, incremento del olor corporal, aparición de acné en áreas seborreicas y sensación subjetiva de aceleración de la velocidad de crecimiento. No constata, sin embargo, aparición de axilarquia ni telarquia. Antecedentes personales y familiares, sin interés.
Exploración física: peso 13,4 kg (P75); talla 88 cm (P97); presión arterial imposible de medir por ausencia de colaboración. Acné pustuloso en frente y barbilla. No se observa hirsutismo, axilarquia ni telarquia. Los genitales son femeninos destacando una pubarquia grado II de Tanner con clitorimegalia (fig. 1). Resto sin alteraciones.

Figura 1. Genitales de la paciente. Pubarquia y clitoromegalia.
Exploraciones complementarias: ante los hallazgos de la exploración física y la rápida instauración de los síntomas se realiza, con carácter urgente, una radiografía de muñeca izquierda que muestra un edad ósea de 3 años y 6 meses (adelanto de 1,5 año), y una ecografía pélvica/ suprarrenal que detecta, en el lugar de la suprarrenal izquierda, una masa sólida de ecogenicidad homogénea y 3,5 × 2,8 cm de tamaño.
Preguntas
¿Cuál es su sospecha diagnóstica?, ¿qué pruebas solicitaría a continuación?
Neoplasia adrenocortical productora de andrógenos
Ante la sospecha de neoplasia adrenocortical productora de andrógenos se solicitan hormona adrenocorticotropa (ACTH) 20 pg/ml (valor normal [VN]: 10-55 pg/ml), cortisol basal plasmático 8,9 µg/dl (VN: 7-26 µg/dl), testosterona 2,5 ng/ml (VN = < 1 ng/ml), deshidroepiandrostendiona sulfato (SDHEA) 615 µg/dl (VN: 82-300 µg/dl). Con objeto de descartar el neuroblastoma así como la existencia de metástasis, se realizan catecolaminas en sangre y orina (normales) y una tomografía computarizada (TC) que confirma los hallazgos ecográficos sin encontrarse invasión aparente de áreas vecinas ni metástasis en hígado o pulmón (fig. 2).
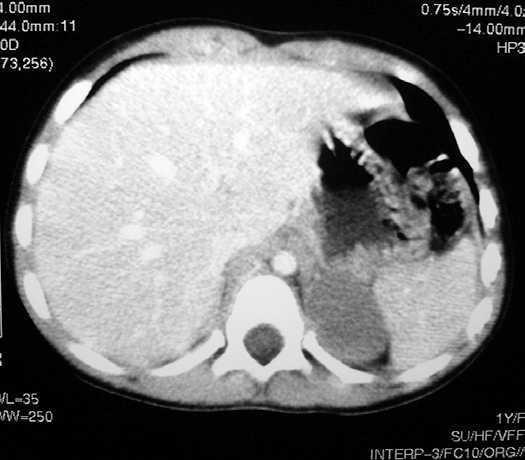
Figura 2.TC toraco-abdominal.
La anatomía patológica de la pieza extirpada cumplía criterios histológicos de carcinoma suprarrenal. Ante la inexistencia de metástasis se optó, en lo sucesivo, por un seguimiento hormonal cada 3 meses durante los primeros 2 años, acompañado de control ecográfico semestral o si se evidencian alteraciones analíticas. Si la paciente no presenta recidiva pueden distanciarse las revisiones con carácter semestral y control ecográfico anual.
Los tumores de la corteza adrenal son raros en la infancia habiéndose calculado una incidencia de 3:1.000.000 en pacientes menores de 20 años1. Suelen afectar a niños con una edad inferior a 5 años1-3 y predominan en el sexo femenino con una proporción 2-3:11,4,5.
Estos tumores asientan preferentemente en niños sin patología subyacente. El cuadro clínico depende de la secreción hormonal. Un alto porcentaje de los casos presentan clínica derivada de un estado de hiperandrogenismo: clitorimegalia en la niña y macrogenitosomía en el varón, asociados a pubarquia, hirsutismo, acné, aceleración del crecimiento y la edad ósea, desarrollo muscular excesivo, etc.1,4. A veces asocian síntomas de hipercortisonismo como obesidad e hipertensión. La aparición de un síndrome de Cushing puro es excepcional2,4, así como también lo es el de hiperestrogenismo1,4 (ginecomastia en el niño y telarquia precoz en la niña) y el de hiperaldosteronismo (hipertensión arterial)1,2,6-8; sólo un 8 % son afuncionantes1,5,8. En un 10 % de los casos puede palparse la masa tumoral, pudiendo ser en algunos casos el primer síntoma de la enfermedad4.
Debe alertar al clínico la rápida progresión de los síntomas anteriormente mencionados, esencialmente los derivados del hiperandrogenismo1. Se han utilizado unos niveles elevados de testosterona tres veces superior a la normalidad (> 2 ng/ml) o los de DHEA-S más allá de 300 µg/dl como guía para iniciar un estudio en busca de un tumor ovárico o suprarrenal como origen del exceso de andrógenos1. En este sentido, la ecografía abdominal en busca de masa suprarrenal y ovárica debe constituir la prueba de primera elección4.
Desde el punto de vista anatomopatológico, la rareza de estos tumores ha obligado a la adopción, a lo largo de los años, de diversos criterios diagnósticos capaces de diferenciar entre adenoma y carcinoma. Un carcinoma podrá sospecharse ante la existencia de una masa con cápsula hiperecoica y focos ecoicos en el interior así como por un tamaño superior a 3 cm; los adenomas, por el contrario, suelen ser más pequeños2. Es frecuente encontrar atrofia del resto del córtex e, incluso, de la suprarrenal contralateral2.
La TC y/o resonancia magnética (RM) desempeñan un papel fundamental en cuanto a una mejor aproximación sobre el tipo de tumor así como su extensión4.
El tratamiento en ambos casos, adenomas y carcinomas, es la exéresis quirúrgica del tumor que resulta curativa para los primeros y, si no hubiera metástasis, también para los segundos1,2,4. Durante la intervención quirúrgica será preciso, si hubiera atrofia suprarrenal contralateral o supresión del eje, un tratamiento con corticoides a dosis de estrés que posteriormente habrá que descender paulatinamente4. Tras la intervención podría ser de utilidad una nueva medición hormonal que orientaría sobre la posibilidad de metástasis ocultas1. En caso de metástasis puede ser necesario el empleo de mitotane y radioterapia aunque los resultados son malos siendo la supervivencia, hoy día, escasa4.
Correspondencia: Dra. A. Jordán Jiménez.
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz.
P.º Castellana, 261. 28046 Madrid. España.
Correo electrónico: africajordan@hotmail.com
Recibido en marzo de 2006.
Aceptado para su publicación en septiembre de 2006.
