



Caso clínico
Recién nacido a término varón, de peso adecuado (38 semanas, 2.680 g). Madre, 29 años, primigesta. Gestación controlada. Serologías de infección connatal: rubéola inmune; toxoplasma, lúes, virus de la inmunodeficiencia humana (VIH), hepatitis B: negativos. Exudado vaginorrectal para estreptococo del grupo B negativo a las 36 semanas de gestación. Ecografías prenatales normales. Resto de antecedentes familiares y personales, sin interés.
Nace tras cesárea urgente por riesgo de pérdida de bienestar fetal. Presentación cefálica. Amniorrexis intraparto. Líquido ligeramente teñido de meconio. Apgar 9/10, reanimación superficial. pH de cordón: 7,24-7,28. Tras el nacimiento comienza con dificultad respiratoria con quejido, aleteo nasal, tiraje subcostal y polipnea de 80-90 resp./min (Silverman: 4-5), iniciándose oxigenoterapia suplementaria indirecta (máximo 40 % a las 12 h de vida). En la exploración física se aprecia un recién nacido vital, con buen estado general, normocoloreado, y con buena perfusión periférica. Temperatura: 36,8 °C. Presión arterial: normal. Auscultación cardíaca: latido cardíaco desplazado a la derecha. Soplo pansistólico, áspero II-III/VI en mesocardio no irradiado. Pulsos periféricos palpables simétricamente. Abdomen blando, depresible, sin masas ni megalias. Resto de exploración física: sin hallazgos.
Con este cuadro clínico, ante la presencia de dificultad respiratoria inmediata y desplazamiento del latido hacia la derecha, se realiza radiografía de tórax urgente (fig. 1) apreciándose una opacificación en hemitórax derecho, sin visualizarse claramente la silueta cardíaca, no objetivándose neumotórax. El pulmón izquierdo parece mostrar una hiperinsuflación compensadora. Se realizan además: hemograma: normal. Gasometría capilar: mínima acidosis respiratoria. Hemocultivo: negativo. Ecografías abdominal y craneal: normales. Ecocardiografía: situs solitus. Dextroposición con levoápex, comunicación interauricular amplia con cortocircuito izquierda-derecha, comunicación interventricular perimembranosa de 4-5 mm tapada por tejido accesorio de la válvula tricúspide, ductus arterioso persistente con cortocircuito izquierda-derecha. Cariotipo: masculino normal (46XY).
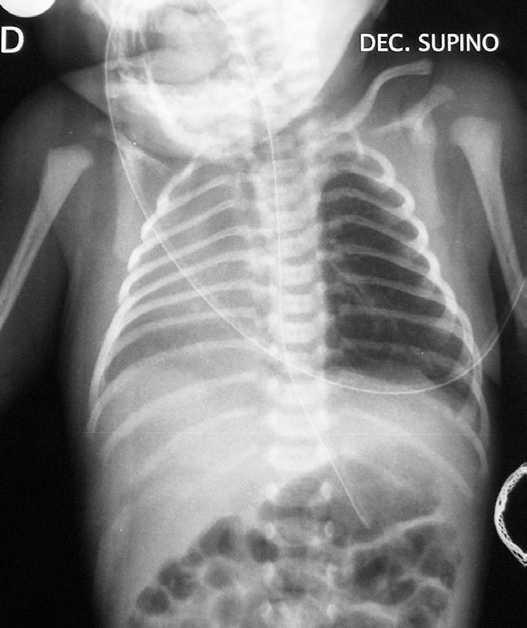
Figura 1. Radiografía simple de tórax: opacificación de hemitórax derecho. Hiperinsuflación compensadora de pulmón izquierdo. No evidencia de aire ectópico.
La evolución del niño es favorable en las siguientes horas, cediendo progresivamente la dificultad respiratoria, y manteniendo buenas saturaciones sin necesidad de oxígeno suplementario a partir del primer día de vida. Con este cuadro clínico se realiza tomografía computarizada (TC) torácica con contraste (figs. 2 y 3) para completar el estudio.
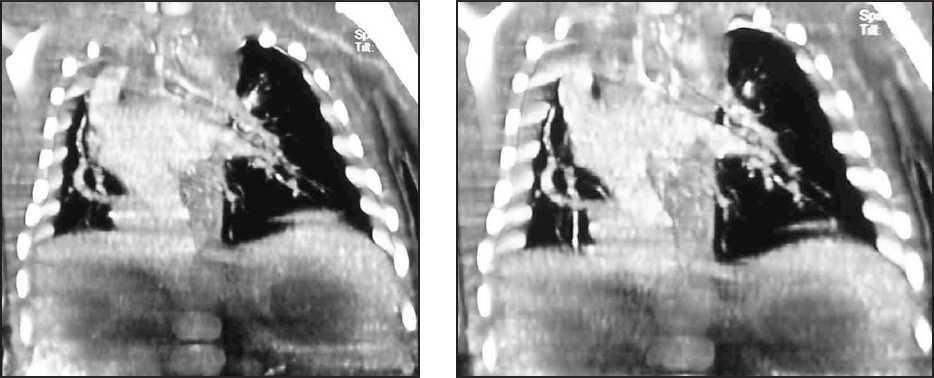
Figura 2.TC torácica con contraste.
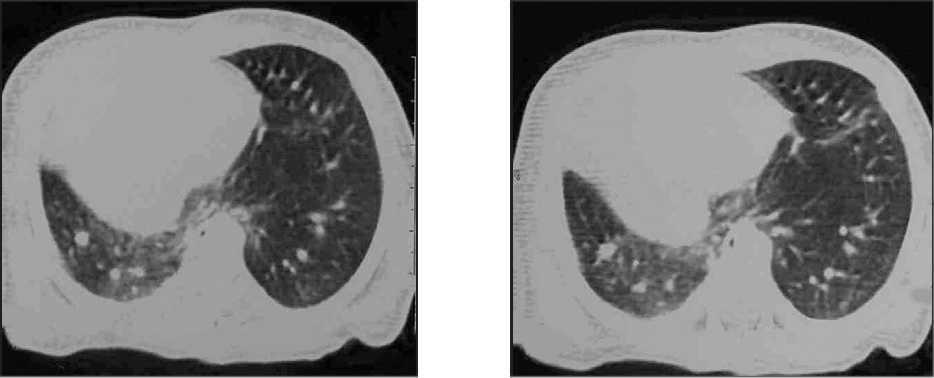
Figura 3.TC torácica con contraste.
Pregunta
¿Cuál es su diagnóstico?
Diagnóstico
Síndrome venolobular congénito con pulmón en herradura.
Comentarios
El término síndrome venolobular congénito (descrito por primera vez por Chassinat en 1836)1 se utiliza para designar un amplio espectro de alteraciones en el desarrollo pulmonar que pueden aparecer de manera aislada o en combinación. En su forma completa consiste en una hipoplasia del pulmón derecho hay pocos casos descritos con afectación izquierda dextrocardia, drenaje venoso anómalo pulmonar derecho (parcial o total) a la vena cava, hipoplasia de la arteria pulmonar derecha, e irrigación arterial anómala del lóbulo inferior derecho desde la aorta torácica o abdominal2,3. Este síndrome también se asocia con otras alteraciones anatómicas (bronquiales, diafragmáticas, vertebrales), e incluso con alteraciones del tracto genitourinario. Históricamente ha recibido otros nombres como síndrome del pulmón hipogenético, síndrome de la vena cava broncovascular o síndrome de la cimitarra4,5. Esta última denominación viene dada por la forma radiológica que en ocasiones presenta el drenaje venoso anómalo del pulmón a la vena cava inferior, a lo largo del borde cardíaco derecho, con una forma semejante a dicha espada turca, y que muchas veces es un hallazgo radiológico casual, diagnosticado generalmente en adultos jóvenes.
La presentación clínica de esta entidad es muy variable, desde niños asintomáticos, hasta otros con insuficiencia cardíaca e hipertensión pulmonar grave. Los recién nacidos suelen tener síntomas más graves que los niños mayores, y es frecuente que presenten malformaciones intratorácias o extratorácicas que condicionan el pronóstico. Las malformaciones que se asocian más frecuentemente (hasta en un 73 %) suelen ser cardíacas (alteraciones de los tabiques ventriculares, estenosis pulmonar, coartación de aorta, etc.)1-5. También pueden aparecer otras alteraciones aún más raras y excepcionales (quistes diafragmáticos, pulmón esofágico o gástrico, ausencia de pericardio). La presencia de un pulmón en herradura es una malformación asociada infrecuente, descrita por primera vez en 19626, y se manifiesta clásicamente por la presencia de un istmo de tejido pulmonar que cruza la línea media por detrás del pericardio, conectando los segmentos posterobasales de ambos pulmones7, como así ocurría en nuestro paciente.
La presentación neonatal es rara, y puede ser sospechada de forma antenatal por la presencia en las ecografías fetales de dextrocardia y estrechamiento de la arteria pulmonar derecha en ausencia de hernia diafragmática congénita8. Sin embargo los diagnósticos prenatales de esta entidad son excepcionales, probablemente por la rareza del cuadro y la dificultad en la visualización de los vasos pulmonares aberrantes, debido a su escaso tamaño y la proximidad al corazón y vasos prevertebrales9.
La evolución clínica de nuestro paciente fue favorable en el período neonatal inmediato, manteniéndose asintomático, aunque en los primeros meses de vida desarrolló una hipertensión pulmonar significativa que obligó a un tratamiento médico y cateterismo con embolización de las arterias nutricias del segmento pulmonar aberrante, permaneciendo posteriormente estable.
Correspondencia: Dr. R. Ortiz Movilla.
Servicio de Pediatría. Hospital Universitario de Getafe.
Ctra. de Toledo, km. 12,500. 28905 Getafe. Madrid. España.
Correo electrónico: rortizmovilla@telefonica.net
Recibido en mayo de 2006.
Aceptado para su publicación en octubre de 2006.
