





La enfermedad Niemann-Pick, en el tipo A (NPA, MIM#257100) y en el tipo B (NPB, MIM#670616), es un trastorno genético hereditario de depósito lisosomal causado por una actividad deficiente de la enzima esfingomielinasa ácida (ASM, por sus siglas en inglés; EC 3.1.4.12). La falta de actividad enzimática normal favorece la acumulación de esfingomielina y otros lípidos en los lisosomas, principalmente en las células del sistema fagocítico mononuclear y en las células ganglionares del sistema nervioso central, lo que ocasiona problemas de salud1. El diagnóstico se realiza mediante la determinación de la actividad de la ASM en linfocitos periféricos o fibroblastos cultivados2.
El déficit de ASM afecta a individuos en todo el mundo, y se estima que su prevalencia global es de 0,5-1/100.0003,4. Por lo general, la morbilidad grave en la NPB se asocia con visceromegalia y deterioro neurológico grave4-6.
La enfermedad de Niemann-Pick es un padecimiento autosómico recesivo causado por mutaciones en el gen SMPD1 (MIM*607608). Hasta la fecha se han identificado más de 120 mutaciones causantes del déficit de ASM7. El espectro clínico de la enfermedad de Niemann-Pick es muy amplio, con una heterogeneidad considerable en la edad de inicio, evolución, gravedad y momento de aparición de las manifestaciones clínicas. La NPA es una enfermedad neurodegenerativa de curso rápido y evolución fatal, que se caracteriza por hepatoesplenomegalia masiva, mientras que la NPB no conlleva afección neurológica y se caracteriza, principalmente, por la hepatoesplenomegalia, un perfil lipídico aterogénico e infiltración pulmonar intersticial5,6. Recientemente se han reportado formas atípicas de enfermedad de gravedad intermedia con inicio tardío y una progresión más lenta de la afección neurológica y visceral5. La mayor incidencia del tipo NPB se da en individuos de origen turco, árabe y norafricano3. La mortalidad global de la NPB en la población pediátrica es del 19%5.
El primer caso corresponde a una niña de origen español de cinco años de edad (paciente uno) que presentó sangrado persistente tras ser sometida a una adenoidectomía, y sufrió una parada cardiaca tras la extubación. La valoración preoperatoria reveló una prolongación leve del tiempo de tromboplastina parcial activada (40″) (TTPA) corregida en el estudio de mezcla (33,9″), trombocitopenia con coagulación normal, hepatoesplenomegalia y un patrón reticular en la radiografía de tórax indicativo de infiltración pulmonar (tabla 1).
Parámetros clínicos y bioquímicos
| Parámetros clínicos y bioquímicos | Paciente 1 | Paciente 2 |
|---|---|---|
| Espirometría | Normal | Normal |
| Electrocardiograma | Normal | Normal |
| Ecocardiograma | Normal | Normal |
| Alteraciones hematológicas | Disfunción plaquetaria, ferropenia leve | Disfunción plaquetaria, ferropenia leve |
| Triglicéridos | 130 mg/dL | 175 mg/dL |
| Colesterol total | 209 mg/dL | 237 mg/dL |
| c-HDL | 20 mg/dL | 16 mg/dL |
| c-LDL | 163 mg/dL | 186 mg/dL |
| AST | 74 U/L | 151 U/L |
| ALT | 45 U/L | 114 U/L |
| Radiografía de esqueleto | Normal | Normal |
| Afección neurológica | No | No |
| Manchas en la retina | No | No |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; c-HDL: colesterol-lipoproteína de alta densidad; c-LDL, colesterol-lipoproteína de baja densidad.
La paciente era la hija primogénita de unos padres sanos sin consanguinidad, y no hubo complicaciones durante el embarazo. A partir de los dos años desarrolló insuficiencia respiratoria causada por obstrucción nasal por hipertrofia adenoidea. El sangrado abundante y grave es un signo importante indicativo de NPB y puede marcar el inicio de la enfermedad.
El segundo caso (paciente dos) es el hermano de la paciente uno, de dos años y cinco meses de edad, sin antecedentes personales de interés. No obstante, durante una evaluación rutinaria se detectó una prolongación leve del TTPA (40,2″) corregida en el estudio de mezcla (34,4″), asociando una coagulación normal, hepatoesplenomegalia, distensión abdominal y patrón reticular en la radiografía de tórax indicativo de infiltración pulmonar. El paciente estaba asintomático (tabla 1). En una evaluación posterior de ambos hermanos, no se objetivó déficit de factores, trombocitopenia o alteraciones en la función plaquetaria que pudieran explicar esta complicación.
Es importante destacar que se han descrito varios casos en la literatura de muerte materna por hemorragia posparto8, en los que los principales factores involucrados en el sangrado excesivo, además de la trombocitopenia, eran trastornos de la función hepática, la afectación medular y esplenomegalia. La morbilidad grave en la enfermedad de Niemann-Pick se debe a la visceromegalia y al deterioro neurológico graves, observados en el 8% de los pacientes6. Durante su evolución, los pacientes pueden desarrollar leucopenia, trombocitopenia y trastornos de coagulación progresivos, lo que aumenta el riesgo de infección y hemorragia grave4,5.
El diagnóstico bioquímico de la NPB se realizó midiendo la actividad de la ASM mediante ensayo fluorimétrico, empleándose como substrato el 6-hexadecanoilamino-4-metilumbeliferil fosforilcolina (Carbosynth Ltd)2. También se realizaron otras pruebas bioquímicas centradas en otros biomarcadores: actividad de la quitotriosidasa (QT) mediante ensayo fluorimétrico empleando 4-metilumbeliferil β-D-N,N′,N′′-triacetilquitotriosido (Sigma-Aldrich; EE.UU.) como substrato, tal y como se ha descrito previamente9, determinación de la concentración en sangre de quimiocina ligando 18/quimiocina pulmonar regulada por la activación (CCL18/PARC) mediante ELISA (R&D Systems Europe, Ltd; Reino Unido) como se ha descrito previamente, y medición del 7-cetocolesterol (7-CC) mediante cromatografía líquida y espectrometría de masas en tándem, según el método descrito por Baila-Rueda et al. con ligeras modificaciones. Se interpretó la actividad de la QT con base en la presencia o ausencia de alelo nulo NM_001256125.2:c.1049_1072dup24 en el gen CHIT1 (MIM*600031). Por último, se aisló el ADN de muestras de sangre en EDTA por el método habitual. Se amplificaron la secuencia promotora, los seis exones, casi todos los intrones y la región no traducida 3’ del gen SMPD1 en cuatro fragmentos superpuestos mediante reacción de polimerasa en cadena empleándose pares de cebadores específicos. Se realizó secuenciación bidireccional por el método de Sanger (BigDye® Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems®, EE.UU.) y electroforesis capilar en un analizador ABI 3500XL (Applied Biosystems®, EE.UU.). Se empleó el programa de predicción MutationTaster10 para estimar la probabilidad de que la mutación en cuestión fuera patogénica o una variante neutral.
En ambos pacientes el nivel de actividad de la ASM se encontraba por debajo de 30% (paciente 1: 0,081 nmol/mg prot/h, inhibida en presencia de lisoesfiengomielina hasta 0,013 nmol/mg prot/h; paciente 2: 0,073 nmol/mg prot/h inhibida en presencia de lisoesfiengomielina hasta 0,003 nmol/mg prot/h). Se observó elevación de los marcadores bioquímicos (CCL18/PARC, 7-CC, QT). Por último, las pruebas genéticas revelaron un genotipo heterocigoto compuesto del gen SMPD1 con las mutaciones NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*) y NM_000543.5:c.1096delG (NP_000534.3:p.Phe368Serfs*17) (tabla 2).
Diagnóstico enzimático y molecular. Se observó elevación de biomarcadores en ambos pacientes (CCL18/PARC, 7-CC, QT) con nivel de actividad de la ASM inferior al 30% del observado en el control. Genotipos idénticos en el gen SMPD1 con las mutaciones p.Trp32* (madre) y p.Phe368Serfs*17 (padre) en heterocigosis. El paciente dos y la madre eran portadores de la variante c.1049_1072dup24 en el gen CHIT1
| Diagnóstico enzimático y molecular | Paciente 1 | Paciente 2 | Madre | Padre | Control |
|---|---|---|---|---|---|
| CCL18/PARC (ng/mL) | 1039 | 727 | 12-165 | ||
| 7-CC(ng/mL) | 400,8 | 432,5 | < Límite detección | < Límite detección | 3,5-52,0 |
| Quitotriosidasa (QT) (mol/mL/h) | 607 | 298 | 16 | 63 | 4-133 |
| Actividad ASM (% normal) | 29% | 26% | 70% | 56% | 100% |
| Genotipo QTc.1049_1072dup24 in CHIT1 | Negativo | Heterocigoto | Heterocigoto | Negativo | |
| Genotipo SMPD1 | [p.Trp32*][p.Phe368Serfs*17] | [p.Trp32*][p.Phe368Serfs*17] | p.Trp32*Heterocigoto | p.Phe368Serfs*17Heterocigoto |
ASM: esfingomielinasa ácida; CCL18/PARC: quimiocina ligando 18/quimiocina pulmonar regulada por la activación; QT: quitotriosidasa; 7-CC: 7-cetocolesterol.
Los padres se consideraron portadores con niveles normales de los biomarcadores. La madre de los pacientes era portadora del alelo NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*) y tenía un nivel de actividad de la ASM del 70%. Además, el padre era portador del alelo NM_000543.5:c.1096delG (NP_000534.3:=p.Phe368Serfs*17) y tenía un nivel de actividad de la ASM del 56%. El paciente dos y su madre tenían la variante NM_001256125.2:c.1049_1072dup24 en heterocigosis en el gen CHIT1.
El estudio genético familiar (fig. 1) confirmó que ninguna de las abuelas eran portadoras de las mutaciones detectadas, por lo tanto, los dos abuelos, ambos fallecidos en el momento del estudio, eran los portadores de las mutaciones.
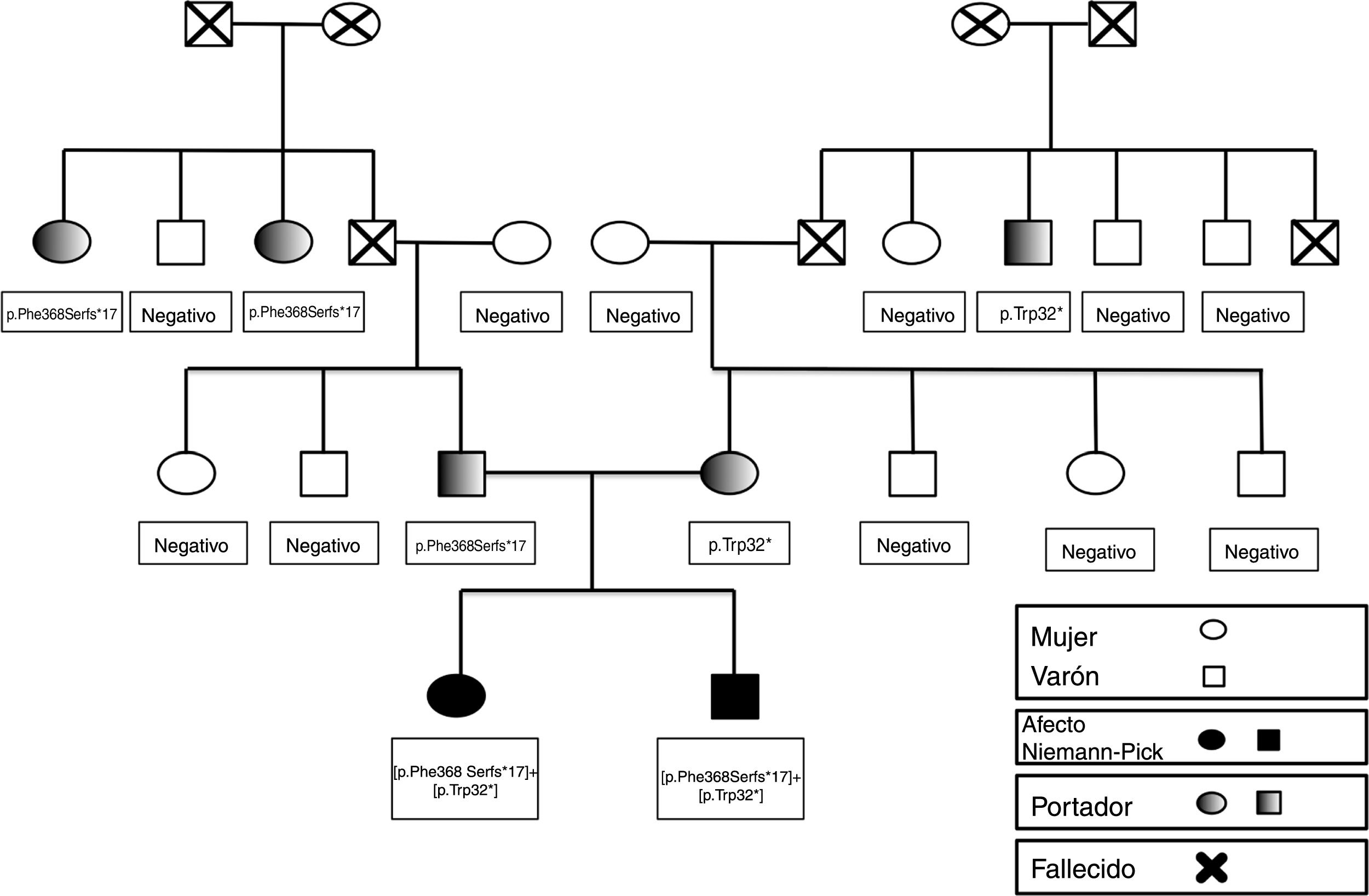
Estudio de portadores familiar: como se expone en el genograma, ninguna de las abuelas son portadoras por lo que ambos abuelos, ya fallecidos, eran los probables portadores de las respectivas mutaciones, que se hallaron en un hermano vivo de cada uno. Ningún otro miembro de la familia está afecto ni existen antecedentes previos.
Las mutaciones en el gen SMPD1 suelen ser privadas y heterocigotas, lo que dificulta el establecimiento de correlaciones genotipo-fenotipo. Aun así, es importante tener en cuenta la posibilidad de asociar mutaciones concretas en el gen SMPD1 con grupos demográficos o étnicos específicos. En cualquier caso, estas mutaciones pueden producir fenotipos leves, intermedios o graves3.
En general, el fenotipo y el nivel de actividad enzimática residual no se asocian con mutaciones específicas. En algunos casos, pacientes con genotipos idénticos tienen fenotipos claramente diferenciados11. Hay datos que sugieren que parte de la heterogeneidad clínica observada en la enfermedad de Niemann-Pick se puede atribuir al fenómeno de impronta paterna, con expresión preferente del alelo materno. Esto también explicaría por qué algunos portadores exhiben una reducción considerable en la actividad de la esfingomielinasa y algunos síntomas de enfermedad de Niemann-Pick11. También hay factores inflamatorios que podrían influir en el nivel de acumulación en vísceras o en las neuronas.12 Por lo tanto, los distintos fenotipos de la enfermedad no se pueden explicar exclusivamente con base en las mutaciones.
Se observaron niveles elevados de los biomarcadores en ambos hermanos (tabla 1), en concordancia con lo descrito por Irun et al12. En cuanto a las variantes del SMPD1 identificadas en esta familia, la mutación NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*), presente en la madre, fue descrita por primera vez por Pittis et al. en pacientes de la región mediterránea13. En dicho estudio, caracterizaron esta mutación como correlacionada con el fenotipo NPB sin afección neurológica, y reportaron su asociación con un alelo previamente descrito en contexto de la NPA. No obstante, también hay descripciones de casos que divergen de este patrón general13.
La mutación NM_000543.5:c.1096delG (NP_000534.3:p.Phe368Serfs*17), detectada en el padre, no se ha descrito anteriormente en la literatura en relación con la enfermedad de Niemann-Pick. De acuerdo con las guías del American College of Medical Genetics and Genomics para la interpretación de variantes, esta mutación se consideraría patogénica (criterios PVS1, PM1, PM2, PM3). Concluimos, por tanto, que la variante NM_000543.5:c.1096delG (NP_000534.3:p.Phe368Serfs*17) era causante de enfermedad.
En ambos casos, la mutación NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*), asociada con un alelo más grave (que podría haber originado la nueva mutación), produjo un fenotipo leve.
El algoritmo diagnóstico propuesto y aplicado en este estudio fue relevante dadas las manifestaciones clínicas, el nivel de actividad enzimática y la secuencia genética en ambos hermanos9. La secuenciación reveló la presencia de la enfermedad mediante la detección de dos variantes patogénicas. Además, se llevó a cabo estudio familiar de mutaciones, que confirmó que cada una de estas variantes estaba localizada en un alelo distinto. Por último, el estudio genético confirmó el diagnóstico enzimático.
Aunque el trasplante de médula ósea puede ser una opción terapéutica en el manejo de la NPB, es importante sopesar los riesgos y beneficios de este procedimiento de manera individualizada5. En nuestros pacientes, este procedimiento se descartó por las posibles complicaciones durante y tras el trasplante.
Con la excepción del trasplante de médula ósea, el tratamiento de la NPB es sintomático4-6. Como posible alternativa, la terapia de reemplazo con enzima humana recombinante ha mostrado ser efectiva en un modelo de ratones knockout de ASM y en ensayos clínicos en adultos y niños14-16. Así, esta terapia podría ser beneficiosa en los casos aquí presentados, ya que son candidatos idóneos para esta opción debido a la ausencia de afectación neurológica. Por último, la terapia génica podría considerarse como una alternativa en el futuro dependiendo de los avances en dicho campo.
Con el propósito de prevenir la aparición de casos adicionales en la familia, se recomienda un estudio familiar de mutaciones para identificar portadores en la familia, así como consejo genético para informar sobre el riesgo de tener una descendencia afectada por la enfermedad. En los casos aquí presentados, recomendamos el diagnóstico genético preimplantacional y estudio enzimático prenatal17 para el cribado de enfermedad de Niemann-Pick.
