





Niemann-Pick disease type A (NPD-A, OMIM#257100) and type B (NPD-B, OMIM#670616) are genetic, inherited lysosomal storage disorders caused by a deficient activity of the enzyme acid sphingomyelinase (ASM; EC 3.1.4.12). The lack of normal enzyme activity allows the accumulation of sphingomyelin and other lipids in the lysosomes, mainly in cells of the mononuclear phagocyte system and in ganglion cells of the central nervous system, causing health problems.1 The diagnosis of NPD is made by measuring the level of ASM activity in peripheral blood lymphocytes or cultured skin fibroblasts.2
Acid sphingomyelinase deficiency (ASMD) affects individuals worldwide with an overall estimated prevalence of 0.5–1 case per 100 000 individuals.3,4 Generally speaking, serious morbidity in NPD-B is associated with visceral organ enlargement and severe neurological impairment.4–6
Niemann-Pick disease is an autosomal recessive disorder caused by mutations in the SMPD1 gene (OMIM*607608). To date, more than 120 mutations have been identified that cause ASMD.7 The clinical spectrum of NPD is very broad, with significant variability in the age of onset, course of disease, severity and timing of clinical manifestations. Niemann-Pick disease type A is a rapid, fatal neurodegenerative disorder manifesting with massive hepatosplenomegaly, whereas type B does not affect the nervous system and is mainly characterized by hepatosplenomegaly, an atherogenic lipid profile and interstitial pulmonary infiltration.5,6 Atypical intermediate forms of disease with a delayed onset and slower neurological and visceral progression have been reported recently.5 The highest incidence of NPD-B is found in people of Turkish, Arabic and North African descent.3 The overall mortality associated with NPD-B in the paediatric population is 19%.5
The first case occurred in a girl aged 5 years of Spanish descent (Patient 1) with persistent bleeding following an adenoidectomy that went into cardiac arrest after extubation. The preoperative evaluation revealed a mildly prolonged activated partial thromboplastin time (40″) (aPTT) that corrected in the mixing study (33.9″), thrombocytopenia with normal coagulation function, hepatosplenomegaly and a reticular lung pattern in the X-ray compatible with pulmonary infiltration (Table 1).
Clinical and biochemical parameters.
| Clinical and biochemical parameters | Patient 1 | Patient 2 |
|---|---|---|
| Spirometry | Normal | Normal |
| Electrocardiogram | Normal | Normal |
| Echocardiogram | Normal | Normal |
| Haematologic abnormalities | Platelet dysfunction, mild iron deficiency | Platelet dysfunction, mild iron deficiency |
| Triglycerides | 130 mg/dL | 175 mg/dL |
| Total cholesterol | 209 mg/dL | 237 mg/dL |
| HDL-c | 20 mg/dL | 16 mg/dL |
| LDL-c | 163 mg/dL | 186 mg/dL |
| AST | 74 U/L | 151 U/L |
| ALT | 45 U/L | 114 U/L |
| Skeletal survey | Normal | Normal |
| Neurologic impairment | None | None |
| Retinal stigmata | None | None |
ALT, alanine aminotransferase; AST, aspartate aminotransferase, HDL-c, high-density lipoprotein cholesterol; LDL-c, low-density lipoprotein cholesterol.
The patient was the firstborn daughter of nonconsanguineous healthy parents, and the pregnancy was uncomplicated. She had respiratory insufficiency due to nasal obstruction caused by adenoidal hypertrophy from age 2 years. Massive and severe bleeding is a significant sign indicative of NPD-B and may represent the onset of disease.
The second case (Patient 2) corresponded to the brother of Patient 1, aged 2 years and 5 months. He did not have any personal history of interest. However, during a routine checkup, we detected a mild prolongation in the aPTT (40.2″) that corrected in the mixing study (34.4″) with normal coagulation function, hepatosplenomegaly, abdominal distension and a reticular lung pattern indicative of pulmonary infiltration in the chest X-ray. He was asymptomatic (Table 1). In a later evaluation of both siblings, we did not find any evidence of factor deficit, thrombocytopenia or platelet function abnormalities that could explain this complication.
For instance, several cases have been described in the literature of maternal death due to postpartum haemorrhage8 in which the main factors behind the bleeding disorders, other than thrombocytopenia, were liver function impairment, bone marrow involvement and splenomegaly. Serious morbidity in NPD involves enlargement of visceral organs and severe neurological impairment, which are present in 8% of these patients.6 In the course of the disease, patients may develop progressive leukopenia, thrombocytopenia and coagulation disorders that increase the risk of infection and severe haemorrhage.4,5
The biochemical diagnosis of NPD-B involved measurement of the level of ASM activity by means of fluorometric enzyme assay, using 6-hexadecanoylamino-4-methylumbelliferyl phosphorylcholine (Carbosynth Ltd) as substrate in leukocytes.2 We also performed other biochemical tests focused on different biomarkers: chitotriosidase activity (ChT) by fluorometric assay using 4-methylumbelliferyl β-d-N,N′,N′′-triacetylchitotrioside (Sigma-Aldrich; USA) as substrate as described previously,9 measurement of serum C-C chemokine ligand 18/pulmonary activation-regulated chemokine (CCL18/PARC) by ELISA (R&D Systems Europe, Ltd, UK) as described elsewhere and measurement of 7-ketocholesterol (7-KC) was quantified using liquid chromatography/tandem mass spectrometry (LC/MS/MS) following the method described by Baila-Rueda et al. with slight modifications. We interpreted the ChT activity based on the presence or absence of the null allele NM_001256125.2:c.1049_1072dup24 in CHIT1 (MIM*600031). Lastly, DNA was isolated from EDTA blood samples using standard methods. The promoter, six exons, almost all intronic regions and the 3´ untranslated region of the SMPD1 gene were amplified in four overlapping fragments by polymerase chain reaction (PCR) using custom primer pairs. We performed bidirectional Sanger sequencing (BigDye™ Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems, USA) and capillary electrophoresis in an ABI 3500XL genetic analyser (Applied Biosystems, USA). We used the Taster10 predictor program to assess the probability of a change being either a pathogenic mutation or a neutral variant.
In both patients, the level of ASM activity was below 30% (patient 1: 0.081nmol/mg protein/h, inhibited in presence of lyso-sphingomyelin to 0.013nmol/mg protein/h; patient 2: 0.073nmol/mg prot/h. inhibited in presence of lyso-sphingomyelin to 0.003nmol/mg protein/h). We observed elevated biomarkers (CCL18/PARC, 7-KC, ChT). Lastly, the genetic study revealed a compound heterozygous SMPD1 genotype for the mutations NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*) and NM_000543.5:c.1096delG (NP_000534.3:=p.Phe368Serfs*17) (Table 2).
Enzymatic and molecular diagnosis. Elevated biomarkers in both patients (CCL18/PARC, 7-KC, ChT) and ASM activity less than 30% of the activity in the control. Identical SMPD1 genotype in both patients, with the mutations p.W32X (mother) and c.1096delG (G367fs*17) (father) in heterozygosis. Patient 2 and his mother had the c.1049_1072dup mutation in heterozygosis in gene CHIT1, which encodes chitotriosidase.
| Enzymatic and molecular diagnostic | Patient 1 | Patient 2 | Mother | Father | Control |
|---|---|---|---|---|---|
| Chemokine CCL18/PARC (ng/mL) | 1039 | 727 | 12−165 | ||
| 7-KC * (ng/mL) | 400.8 | 432.5 | <2 | <2 | 3.5−52 |
| Chitotriosidase (ChT) (mol/mL/h) | 607 | 298 | 16 | 63 | 4−133 |
| ASM activity (% normal) | 29% | 26% | 70% | 56% | 100% |
| ChT genotype c.1049_1072 dup24 in CHIT1 | Negative | Heterozygous | Heterozygous | Negative | |
| SMPD1 genotype | p.W32X + c.1096delG | p.W32X + c.1096delG | pW32X Heterozygous | c.1096delG Heterozygous |
ASM, acid sphingomyelinase; ChT, chitotriosidase.
The parents are considered carriers and have normal biomarker levels. The mother of the siblings carried the NM_000543.5:c.95G>A allele (NP_000534.3:p.Trp32*) and had an ASM activity of 70% of normal. Besides, the father carried the NM_000543.5:c.1096delG allele (NP_000534.3:=p.Phe368Serfs*17) and had an ASM activity of 56% of normal. Patient 2 and his mother were heterozygous for NM_001256125.2:c.1049_1072dup24 in the CHIT1 gene.
Mutation analysis in the family (Fig. 1) confirmed that neither grandmother carried any of the detected mutations. Thus, the 2 grandfathers, deceased at the time of the analysis, were the carriers of the respective mutations.
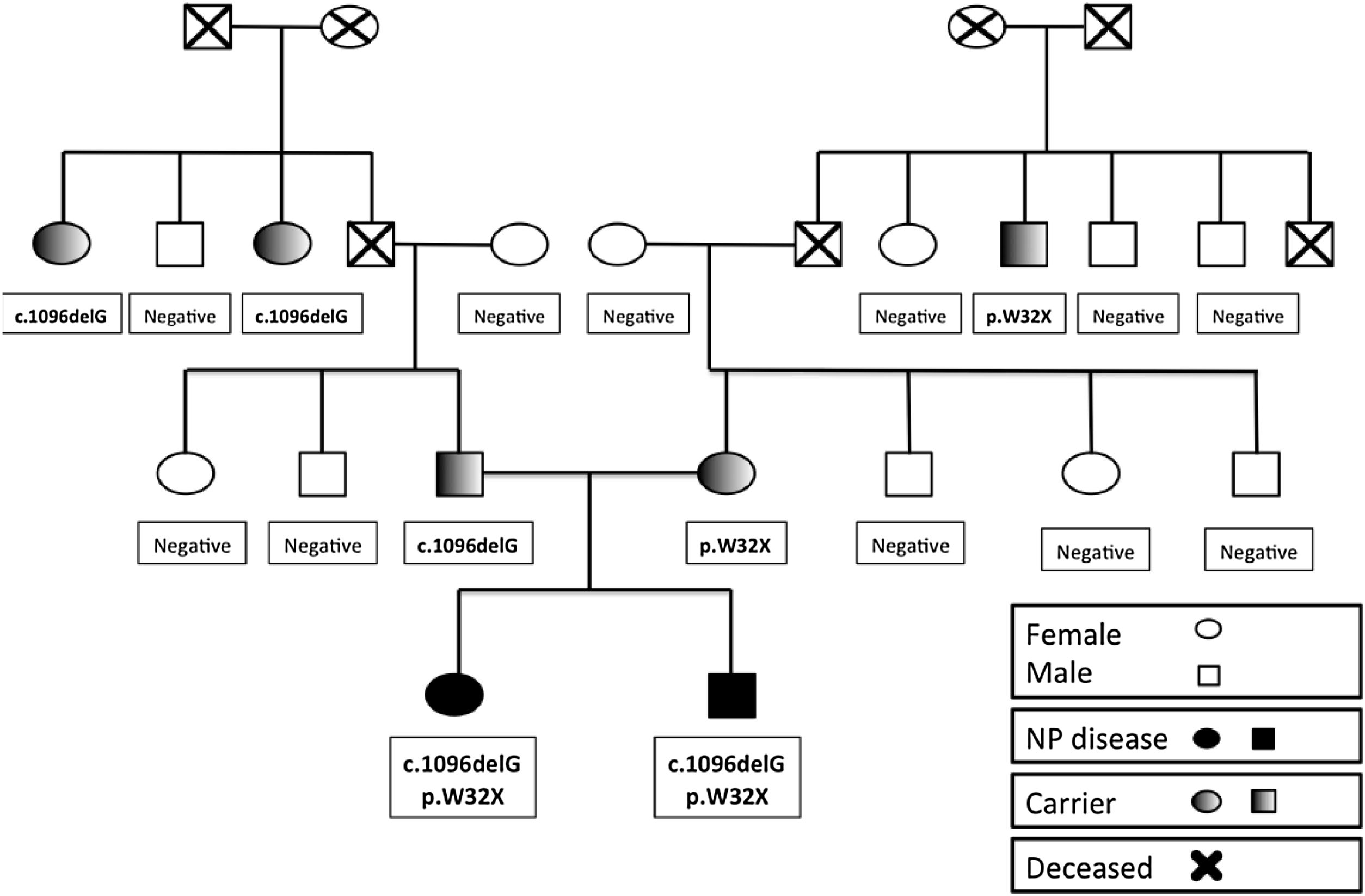
Mutations in the SMPD1 gene are usually private and frequently heterozygous, making it difficult to establish genotype-phenotype correlations. Still, it is also important to consider that specific mutations of the SMPD1 gene may be associated with certain demographic or ethnic groups. All in all, these mutations can produce mild, intermediate or severe phenotypes.3
In general, the phenotype or the level of residual enzyme activity are not associated with specific mutations. Sometimes patients with identical genotypes display distinct phenotypes.11 There is evidence suggesting that some of the observed clinical heterogeneity in NPD could be explained by the imprinting of the paternal allele, with preferential expression of the maternal allele. This would also explain why some carriers exhibit a significant decrease in sphingomyelinase activity and some symptoms of NPD.11 Further, there are inflammatory factors that may have an effect on the levels of visceral or neuronal storage.12 Therefore, mutations alone cannot explain the different disease phenotypes.
We found elevation of biomarkers in both siblings (Table 1), consistent with the findings reported by Irún et al.12 As for the SMPD1 variants found in this family, the NM_000543.5:c.95G>A (NP_000534.3:p.Trp32*) mutation, present in the mother, was first described by Pittis et al. in Mediterranean patients.13 In this study, they characterized this mutation as associated with a NPD B phenotype without neurologic involvement, and, furthermore, that it was associated with an allele previously reported in NPD type A. However, it is also possible to find reports that diverge from this general pattern.13
The NM_000543.5:c.1096delG mutation (NP_000534.3:p.Phe368Serfs*17), detected in the father, has not been described in the previous literature on NPD. Based on the interpretation guidelines of the American College of Medical Genetics and Genomics, this new mutation should be considered pathogenic (PVS1, PM1, PM2, PM3 criteria). Hence, we concluded that the NM_000543.5:c.1096delG variant (NP_000534.3:p.Phe368Serfs*17) could be disease-causing.
In both cases, the NM_000543.5:c.95G>A mutation (NP_000534.3:p.Trp32*), associated with a more severe allele (which could be the origin of the new mutation), caused a mild phenotype.
The previously proposed diagnostic algorithm was relevant in these cases given the clinical features, enzyme activity and gene sequencing results found in each sibling.9 Sequencing established the presence of the disease through the detection of the 2 disease-causing variants. In addition, we carried out a family pedigree study that confirmed that each of these variants was located in a different allele. Last of all, the genetic study confirmed the enzymatic diagnosis.
Although bone marrow transplantation can be used for treatment for NPD-B, it is important to weigh the risks and benefits of this procedure in each particular patient.5 This procedure was ruled out in our patients on account of the adverse effects of potential complications during and after transplantation.
Outside of bone marrow transplantation, the treatment of NPD-B is symptomatic.4–6 Alternatively, replacement therapy with recombinant human enzymes has proven effective in an ASM-knockout mouse model and in clinical trials in adults and children.14–16 In this regard, it is possible that the patients presented in this article could benefit from this therapeutic approach, as the absence of neurologic involvement makes them ideal candidates for this therapy. Last of all, gene therapy could be considered in the future depending on the methodological advances in the field.
To prevent the appearance of further cases in the family, we recommend performance of pedigree analysis to search for carriers in the family as well as genetic counselling to inform of the risk of NPD in the offspring. In the cases presented here, we would recommend preimplantation genetic diagnosis and prenatal enzymatic studies17 to screen for NPD.
Please cite this article as: Villar-Guerra Pd, Reig C, Irún P, Moreno B, Giraldo P, Cebolla JJ. Nueva mutación asociada a enfermedad de Niemann-Pick en dos niños españoles: descripción del genotipo, actividad de la esfingomielinasa, fenotipo y revisión. An Pediatr (Barc). 2020. https://doi.org/10.1016/j.anpedi.2020.03.021
