


Las neuropatías sensitivas autonómicas son parte de las neuropatías periféricas y se deben a disfunción de genes involucrados en el funcionamiento de las neuronas sensoriales y autonómicas. Se conocen 6 variantes clínicas con subtipos determinados por anormalidad en 11 genes, los diferentes fenotipos varían en la edad de inicio, presencia de disautonomías y patrón de herencia, que con excepción del tipo i son autosómicas recesivas. La neuropatía sensitiva autonómica tipo ii se caracteriza por déficit de la sensibilidad al dolor, temperatura y propiocepción. Puede manifestarse al nacer o iniciar entre los 10 y 20 años de edad con úlceras, mutilaciones y amputaciones acrales. En el presente estudio se describe a 3 miembros de una familia con mutación en el gen WNK1 causante de la forma IIA de esta neuropatía hereditaria.
The hereditary sensory and autonomic neuropathies are genetic disorders characterized by the loss of sensation including pain, tactile and temperature. Its clinical and molecular features vary widely; the symptoms may begin from birth or be noticed in the first or second decade, with different types of complications of trauma to the extremities such as ulcers, mutilations and acral amputations. They are classified into six groups from I to VI, determined by the abnormality in eleven genes leading to phenotypic variations in the age of onset and the presence or absence of dysautonomia signs. With the exception of type I, all are autosomal recessive. The type II of these neuropathies is characterized by insensitivity to pain, heat and proprioception. We describe three members of a Mexican family with WNK1 gene mutation that caused hereditary neuropathy IIA.
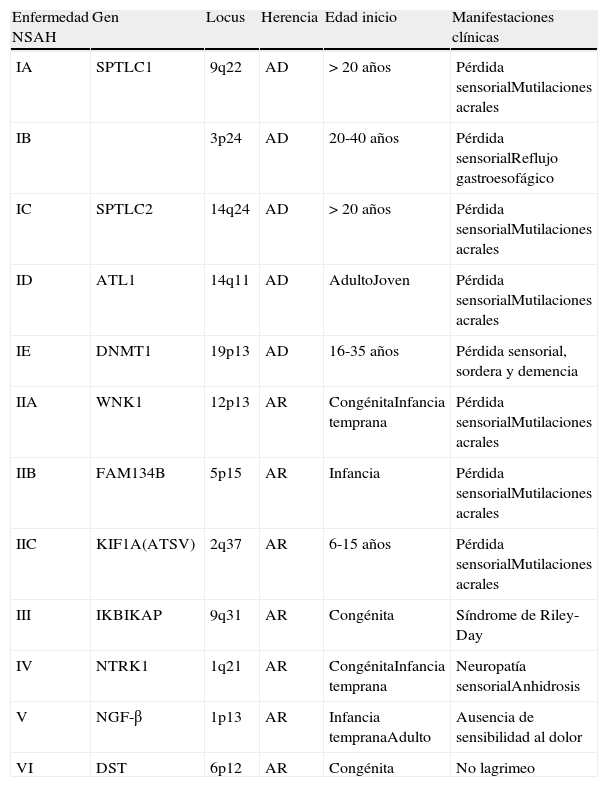
Las neuropatías sensitivas autonómicas hereditarias (NSAH) son desórdenes de causa genética en los que están afectados los axones o el citoesqueleto de las neuronas sensoriales y autonómicas1. Se conocen 6 variantes clínicas con 11 genes causales que varían en la edad de inicio, la presencia de disautonomías y el patrón de herencia que, con excepción del tipo i que es autosómica dominante, el resto se heredan en forma recesiva (tabla 1)2,3.
Clasificación de las neuropatías sensitivas. Se describen los 6 tipos, muestra los patrones de herencia, las subclasificaciones definidas por la anormalidad en genes diferentes y características clínicas que ayudan a la distinción clínica entre las diferentes variedades de insensibilidad hereditaria al dolor
| Enfermedad NSAH | Gen | Locus | Herencia | Edad inicio | Manifestaciones clínicas |
| IA | SPTLC1 | 9q22 | AD | > 20 años | Pérdida sensorialMutilaciones acrales |
| IB | 3p24 | AD | 20-40 años | Pérdida sensorialReflujo gastroesofágico | |
| IC | SPTLC2 | 14q24 | AD | > 20 años | Pérdida sensorialMutilaciones acrales |
| ID | ATL1 | 14q11 | AD | AdultoJoven | Pérdida sensorialMutilaciones acrales |
| IE | DNMT1 | 19p13 | AD | 16-35 años | Pérdida sensorial, sordera y demencia |
| IIA | WNK1 | 12p13 | AR | CongénitaInfancia temprana | Pérdida sensorialMutilaciones acrales |
| IIB | FAM134B | 5p15 | AR | Infancia | Pérdida sensorialMutilaciones acrales |
| IIC | KIF1A(ATSV) | 2q37 | AR | 6-15 años | Pérdida sensorialMutilaciones acrales |
| III | IKBIKAP | 9q31 | AR | Congénita | Síndrome de Riley-Day |
| IV | NTRK1 | 1q21 | AR | CongénitaInfancia temprana | Neuropatía sensorialAnhidrosis |
| V | NGF-β | 1p13 | AR | Infancia tempranaAdulto | Ausencia de sensibilidad al dolor |
| VI | DST | 6p12 | AR | Congénita | No lagrimeo |
La NSAH tipo ii se caracteriza por disminución progresiva de la sensibilidad al tacto, dolor, temperatura y propiocepción; inicia desde el nacimiento o entre la primera y la segunda décadas con la aparición de úlceras, mutilaciones y amputaciones acrales. En ocasiones, hay fracturas, artropatía neuropática con arreflexia y osteomielitis. Las manifestaciones autonómicas no son muy marcadas, predominando la hiperhidrosis, la incontinencia urinaria y la respuesta pupilar lenta4. La NSAH tipo ii es una afección rara, con prevalencia mundial desconocida, y casi el 50% de los casos reportados provienen del este de Canadá5.
En el presente artículo se describen los hallazgos neurológicos, neurofisiológicos y esqueléticos en 3 miembros de una familia con NSAH II.
Pacientes y métodosEl caso índice acudió a los 16 años de edad, con antecedente de padres consanguíneos en segundo grado y neurodesarrollo normal. A los 8 años inició con pérdida gradual de la sensibilidad de la rodilla hasta los dedos de los pies, sin disautonomías; posteriormente desarrolló úlceras en las plantas de ambos pies, nula sensibilidad al traumatismo en los ortejos, con celulitis, necrosis y amputaciones espontáneas indoloras de varios dedos de los pies. El propósito tiene 2 hermanos afectados de 12 y 10 años.
El examen físico con funciones cognitivas íntegras, pares craneales normales, fuerza 5/5 en las extremidades superiores, ausencia de la falange distal del tercer dedo de la mano derecha, en las extremidades inferiores fuerza 5/5 proximal, distal no valorable por presencia de celulitis y úlceras plantares, amputación de primeros ortejos y del quinto dedo izquierdo, pérdida de falanges en el resto de los dedos (fig. 1). Reflejos de estiramiento muscular en los miembros superiores, normales, rotulianos disminuidos y ausencia de aquíleo bilateral. Sensibilidad al dolor, la temperatura, el tacto y la propiocepción normal en las extremidades superiores y el tronco. De la rodilla a los dedos del pie, pérdida de sensibilidad y propiocepción normal. Velocidad de conducción nerviosa con potenciales motores normales y ausencia de potenciales sensitivos. En las úlceras se cultivó Proteus spp.; en la tomografía computarizada (TC) (fig. 2) se observó la ausencia de falanges distales, con datos de osteomielitis, y se inició tratamiento con ciprofloxacino.


Los hermanos también mostraron pérdida de la sensibilidad distal de rodillas a pies, ulceraciones cicatrizadas, sin datos de infección y pérdida de algunas falanges.
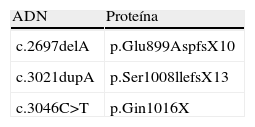
Se realizó una secuenciación de ADN para descartar NSAH II, reportándose una mutación sin sentido en estado homocigoto c.868C>T en el gen WNK1 que cambia una arginina por un codón de paro en la posición 290 de la proteína p.R290X.
DiscusiónEl diagnóstico de NSAH II se fundamenta en la ausencia congénita de sensibilidad al dolor, el tacto y la temperatura, de predominio distal, que se hace evidente en la primera o segunda décadas de la vida. En el inicio se desarrolla una «piel neuropática» seca y gruesa, que favorece el desarrollo de ulceraciones e infecciones. Las fracturas secundarias a la osteomielitis se presentan con la notoria característica de la ausencia de dolor; pueden pasar meses antes de que se considere anormal el carácter recurrente de las lesiones, ocasionando un diagnóstico tardío, con incremento de la morbilidad6.
El desarrollo de úlceras en las manos y los pies, y las amputaciones espontáneas marcan el momento en que se busca atención médica. A la pérdida de sensibilidad la acompaña la pérdida progresiva de los reflejos de estiramiento muscular. A diferencia de otras NSAH, las manifestaciones autonómicas son pocas y el desarrollo cognitivo es normal, lo que la diferencia de las tipos iii y iv6.
Los estudios de neuroconducción y la biopsia de nervio muestran ausencia o disminución de los potenciales sensitivos y motores normales o discretamente reducidos. La biopsia muestra pérdida importante de fibras pequeñas mielinizadas y disminución de las no mielinizadas7,8. Estos procedimientos invasivos pueden ser sustituidos por los estudios moleculares.
La NSAH tipo ii ocurre por mutaciones en 3 genes, WNK1, FAM134B o KIF1A, lo que da lugar a las formas A, B y C de la enfermedad. Los afectados con la forma B tienen disautonomías, mientras que en la forma C muestran anormalidad motora distal. En particular, la forma IIA (OMIM # 201300) se asocia a mutaciones en el gen de la proteína cinasa de treonina/serina (WNK1), localizado en 12q139. La correlación del gen afectado con las manifestaciones clínicas no es contundente, por lo que habitualmente se realizan paneles con varios genes.
El análisis molecular de las NSAH es complejo, aun cuando se estudien varios genes simultáneamente. El estudio sistemático de los genes SPTLC1, RAB7, WNK1/HSN2, FAM134B, NTRK1 y NGFB llevará a identificar la anormalidad genética en el 15 al 20% de los casos. WNK1/HSN2, FAM134B, NTRK1 y NGFB causan las formas recesivas y cuando se toma en cuenta el patrón de herencia la posibilidad de encontrar la mutación responsable se incrementa al 40%, siendo más frecuente en WNK1/HSN2 debido a mutaciones sin sentido o por corrimientos del marco de lectura en el exón 10.
La descripción original de esta afección se realizó en una familia árabe, pero la mayor frecuencia de casos está descrita a inicios del siglo xix en comunidades canadienses de Nueva Escocia, Québec y Terranova5,10,11. Las mutaciones más frecuentes en estos individuos se listan en la tabla 2. Se piensa que son resultado de un efecto fundador por ancestros comunes en Inglaterra. En la actualidad, los casos se distribuyen en todo el mundo.
En un reporte previo de 2 familias mexicanas afectadas de NSAH IIA, se identificó una eliminación nueva de 8 nucleótidos c.1219_1226 delTCTCAGCA, que ocasiona una proteína anormal. Los afectados presentaban ausencia de sensibilidad y amputaciones en las porciones distales de manos y pies, sin debilidad muscular ni manifestaciones autonómicas, potenciales sensitivos ausentes con edad de inicio entre la primera y la segunda décadas de la vida (9-20 años)12. Nuestros casos mostraron un cuadro prácticamente idéntico, con una mutación previamente descrita en un individuo canadiense con ancestros libaneses13.
El origen padecimientote la afección en Latinoamérica se desconoce, pudiendo ser resultado de mutaciones nuevas, como la descrita previamente en México o heredada a través de ancestros comunes. Este caso señala la posibilidad de migración a México de individuos con ancestros libaneses-canadienses o la existencia de un sitio con tasa de mutación superior a la esperada en la secuencia de WNK12,14.
La fisiopatología padecimientote la enfermedad se ha comenzado a conocer; WNK1 codifica isoformas de la proteína WNK1 que participan en el control de la presión arterial y la sensibilidad mediante la regulación de la actividad de canales de sodio y potasio. El flujo de iones en la periferia de las neuronas modula el crecimiento y la maduración de las extensiones neurales de las células de Schwann y el tracto de Lissauer15,16. Por su parte, FAM134B en el locus 5p15.1 (OMIM #613115) produce una proteína estructural del complejo Golgi de las neuronas de ganglios sensoriales; sus formas mutantes producen disfunción en procesos celulares encargados de la supervivencia axonal17,18, mientras que KIF1A en el locus 2q37 (OMIM #614213) produce una proteína de la familia de las kinesinas, involucrada en el transporte axonal anterógrado de WNK119,20.
El conocimiento de la fisiopatología aporta datos valiosos para el futuro desarrollo de tratamientos que eviten el proceso de desmielinización o que protejan la mielina de los axones sensitivos. El tratamiento es sintomático y, sobre todo, preventivo, por lo que es necesaria la participación de un equipo multidisciplinario. El entrenamiento a pacientes y familiares es importante, enseñándoles la autoexploración del pie para la búsqueda de signos de traumatismo. Una de las medidas más importantes es el uso de calzado adecuado y la hidratación de la piel para evitar osteomielitis y amputaciones10.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
