


El síndrome de regresión testicular o anorquia bilateral se define como la ausencia de tejido testicular en sujetos con genitales externos de aspecto masculino y cariotipo 46XY. Tiene una incidencia aproximada de uno de cada 20.000 recién nacidos 46XY, de manera que afecta a menos del 5% de los varones con criptorquidia1,2. Dada la infrecuencia de esta entidad nosológica, a continuación se presenta el siguiente caso clínico:
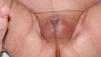
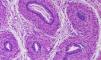
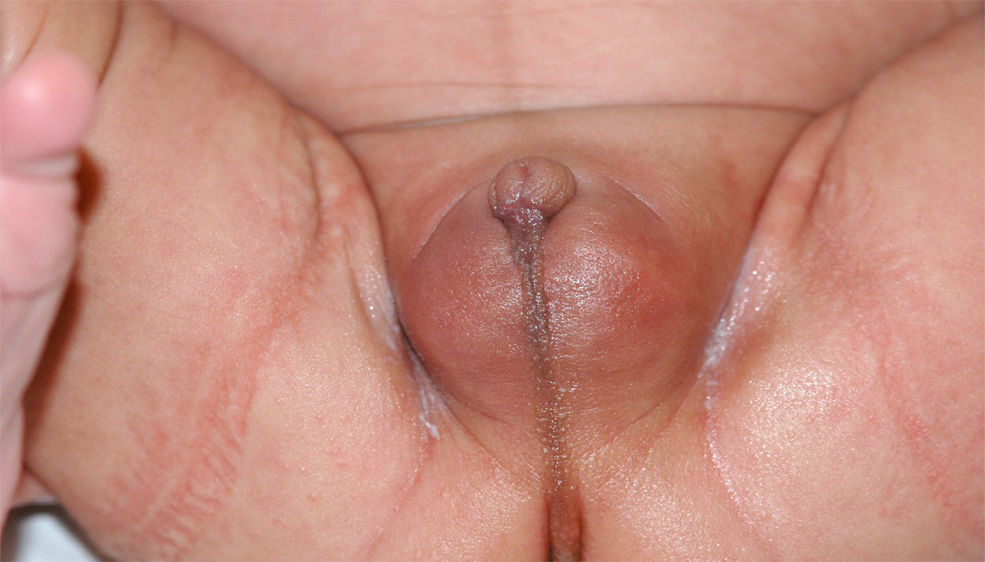
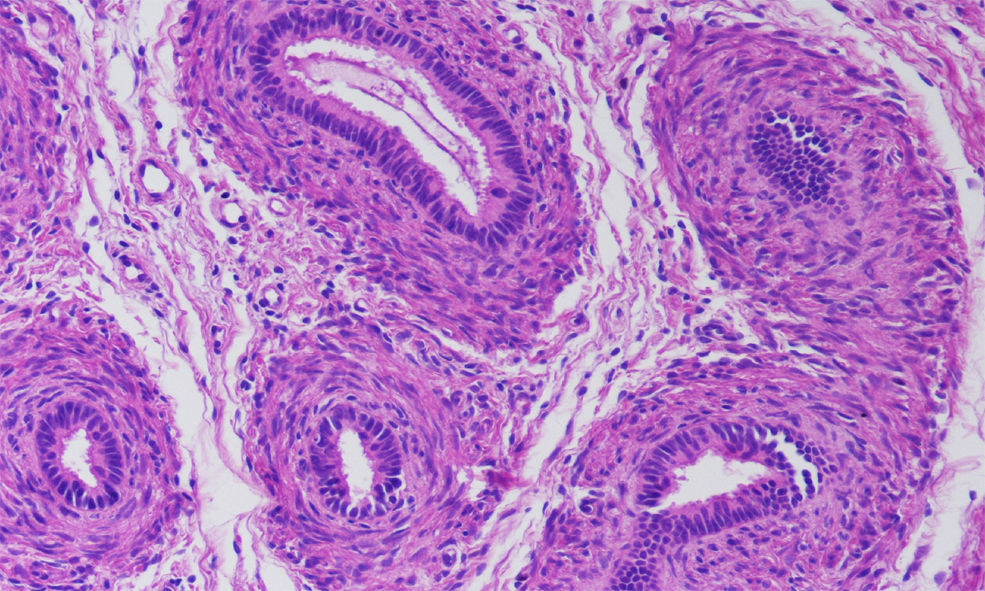
Recién nacido a término, con un peso adecuado para la edad gestacional y una longitud de 49cm (percentil 25), con micropene (0,7×0,2cm), criptorquidia bilateral e hipoplasia escrotal (fig. 1), con padres consanguíneos de 2.° grado y sin otros antecedentes de interés. A las 48 horas de vida se inició el estudio etiológico solicitando un cariotipo que fue 46 XY. La ecografía abdominal mostró 2 estructuras hipoecogénicas mal definidas en los conductos inguinales, sin evidencia de restos mulerianos. Durante la primera semana de vida se le realizó un estudio hormonal del que se obtuvieron los siguientes datos: hormona foliculoestimulante, 12U/l (valores normales [VN]: 0,2–3,5); luteinizante, 17,5U/l (VN: 0,5–6,5); testosterona, 0,3ng/ml (VN: 0,3–2,9); 17-hidroxiprogesterona, 3,09ng/ml (VN: 0,4–10), y hormona antimuleriana (HAM), 0,1ng/ml (VN: 10–43). Por otro lado, se obtuvieron resultados normales tanto de la gasometría venosa como del ionograma. Posteriormente, se realizó un test de estimulación con gonadotropina coriónica humana a dosis de 1.000U/día por vía intramuscular cada 48h3, en 3 dosis en total. La concentración de testosterona se determinó 24h después de la última inyección y fue de 0,45ng/ml (respuesta anómala). Esta falta de síntesis de testosterona podría estar motivada por una alteración enzimática o por un trastorno del desarrollo gonadal. En este punto, la concentración casi indetectable de la hormona antimuleriana (0,1ng/ml) sugirió un trastorno del desarrollo gonadal, cuyo diagnóstico definitivo es la biopsia testicular. Finalmente, se practicó una laparotomía suprapúbica exploradora en la que se realizó la biopsia de los supuestos testículos, localizados en los canales inguinales. En el estudio histológico se identificaron estructuras tubulares con estroma fibromuscular, compatibles con epidídimo (fig. 2).
Hoy en día se especula con 2 teorías que pueden explicar este trastorno. Por un lado, los factores genéticos que contribuyen a una degeneración precoz del tejido testicular. Se sabe la importancia de los genes SRY y SF-1 para la adecuada diferenciación testicular, y que el descenso testicular tiene 2 fases, a saber: a) fase intraabdominal, en la que la participación del factor similar a la insulina tipo 3 es primordial para el desarrollo del gubernáculum, y b) fase inguinal, que depende de los andrógenos4,5. Así, se han estudiado los genes SRY, INSL3 y SF1 en sujetos afectados de anorquia bilateral y se encontró sólo un sujeto con mutación en heterocigosis del gen SF16,7. Por tanto, los argumentos a favor de esta hipótesis incluyen que es una degeneración testicular bilateral, como si se tratara de un proceso apoptótico, y que hay una mayor incidencia familiar. En el sujeto del caso que se presenta, hay un antecedente de consanguinidad, que orienta hacia un trastorno de origen genético; sin embargo, se investigó el gen SRY con resultados negativos.
Por otro lado, la hipótesis mecánica sostiene que cualquier fenómeno obstructivo que altere el descenso testicular normal puede contribuir al desarrollo de la anorquia bilateral. En contra de esta teoría se explica que es un fenómeno bilateral y que, en la mayoría de las exploraciones quirúrgicas, no suele encontrarse ninguna anomalía estructural1,2.
Esta entidad tiene un espectro clínico muy variable y puede manifestarse como criptorquidia bilateral aislada o relacionada con micropene, con hipoplasia escrotal o con ausencia de fusión labioescrotal, dependiendo del momento del desarrollo fetal en el que desaparecieron los testículos1,4.
Tras la sospecha clínica inicial, los datos de laboratorio que orientan al diagnóstico de anorquia son elevación de las gonadotropinas, disminución de la síntesis de testosterona basal y tras estímulo, y la reducción de la concentración de HAM3,8. La determinación de HAM brinda información sobre la funcionalidad de la célula de Sertoli. Así, la concentración indetectable de la hormona en el contexto de la disminución de la síntesis de testosterona, sugiere un trastorno del desarrollo gonadal (anorquia bilateral frente a disgenesia gonadal), cuyo diagnóstico definitivo es la biopsia gonadal9.
El tratamiento depende de la clínica acompañante. En el caso de la presencia de micropene, se recomienda la administración de 3 inyecciones intramusculares de testosterona, a la dosis de 50 a 100mg/m2 cada 4 semanas1,8. Por otro lado, hay controversia en cuanto a la implantación precoz o tardía de prótesis testiculares. La actitud más extendida es la colocación de éstas entre los 4 y los 6 años, con el objeto de favorecer el desarrollo escrotal y evitar repercusiones psicológicas. Posteriormente, se implantarán las prótesis definitivas en la época puberal junto con una terapia hormonal sustitutiva10.
