



La cefalopolisindactilia de Greig es un síndrome autosómico dominante de baja prevalencia causado por mutaciones en el gen GLI3, localizado en 7p14.1 y caracterizado por la tríada clínica de polisindactilia, macrocefalia e hipertelorismo. En aproximadamente el 20% de los casos se detecta una deleción de tamaño variable. Si la deleción es grande y afecta a otros genes además de GLI3, puede aparecer un fenotipo más severo, aceptándose la denominación de cefalopolisindactilia de Greig-síndrome de genes contiguos para estos casos.
Describimos el caso de una niña recién nacida con polisindactilia, hipertelorismo y microcefalia, que presenta una microdeleción en 7p14.1 de 1,5 Mb de origen paterno diagnosticada mediante array-CGH.
Greig cephalopolysyndactyly is a rare autosomic dominant syndrome caused by mutations in GLI3 gene located on cytoband 7p14.1 and characterized by the clinical triad of polysyndactyly, macrocephaly and hypertelorism. In approximately 20% of the cases a deletion of variable size is detected. If deletion is large and affects other genes as well as GLI3, a more severe phenotype is expected. Thus, Greig cephalopolysyndactyly contiguous gene syndrome is a multiple malformation syndrome caused by haploinsufficiency of GLI3 and adjacent genes.
We describe the case of a newborn female with polysyndactyly, hypertelorism and microcephaly and a 1.5 Mb 7p14.1 microdeletion of paternal origin diagnosed by array-CGH.
La cefalopolisindactilia de Greig (GCPS) (OMIM: 175700) es un síndrome de baja prevalencia (estimada en 1-9/106), heredado según un patrón autosómico dominante, pleiotrópico, caracterizado por la tríada clínica de polisindactilia, macrocefalia e hipertelorismo1. Está causado por la haploinsuficiencia del gen GLI3 en 7p14.12 y es alélico con el síndrome de Pallister-Hall (OMIM: 146510) causado por mutaciones en GLI3 que dan lugar a una proteína truncada3. Aproximadamente, en el 20% de los casos descritos se detecta una deleción, de tamaño variable, que afecta a GLI34,5. Cuando la deleción es mayor de 1 Mb e incluye otros genes, el cuadro clínico es más severo, aceptándose el término cefalopolisindactilia de Greig-síndrome de genes contiguos (GCPS-CGS)1,6,7.Presentamos el caso de una recién nacida con una deleción de 1,5 Mb de origen paterno que afecta a GLI3 y otros genes, con un fenotipo compatible con síndrome de genes contiguos-GCPS.
Caso clínicoRecién nacida pretérmino de 36 semanas, pequeña para edad gestacional, que ingresa desde su nacimiento en la unidad de neonatología por bajo peso. Presenta polidactilia bilateral del primer dedo de los pies.
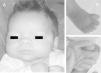
Antecedentes familiares: madre de 31 años, fumadora de unos 20 cigarrillos al día durante el embarazo. Padre con polidactilia del primer dedo de ambos pies (intervenido quirúrgicamente en la infancia), frente amplia y pulgares grandes (fig. 1). En la entrevista refiere dificultades en el aprendizaje escolar, apreciándose en él ligeras alteraciones en el lenguaje. Hermana de 4 años sana, según refieren los padres.
Antecedentes obstétricos: segunda gestación. Embarazo controlado. Restricción del crecimiento intrauterino detectado en el tercer trimestre. Con Doppler normal. Amenaza de parto pretérmino en la semana 34. Parto espontáneo, eutócico. Placenta pequeña (300g). Apgar 9/10.
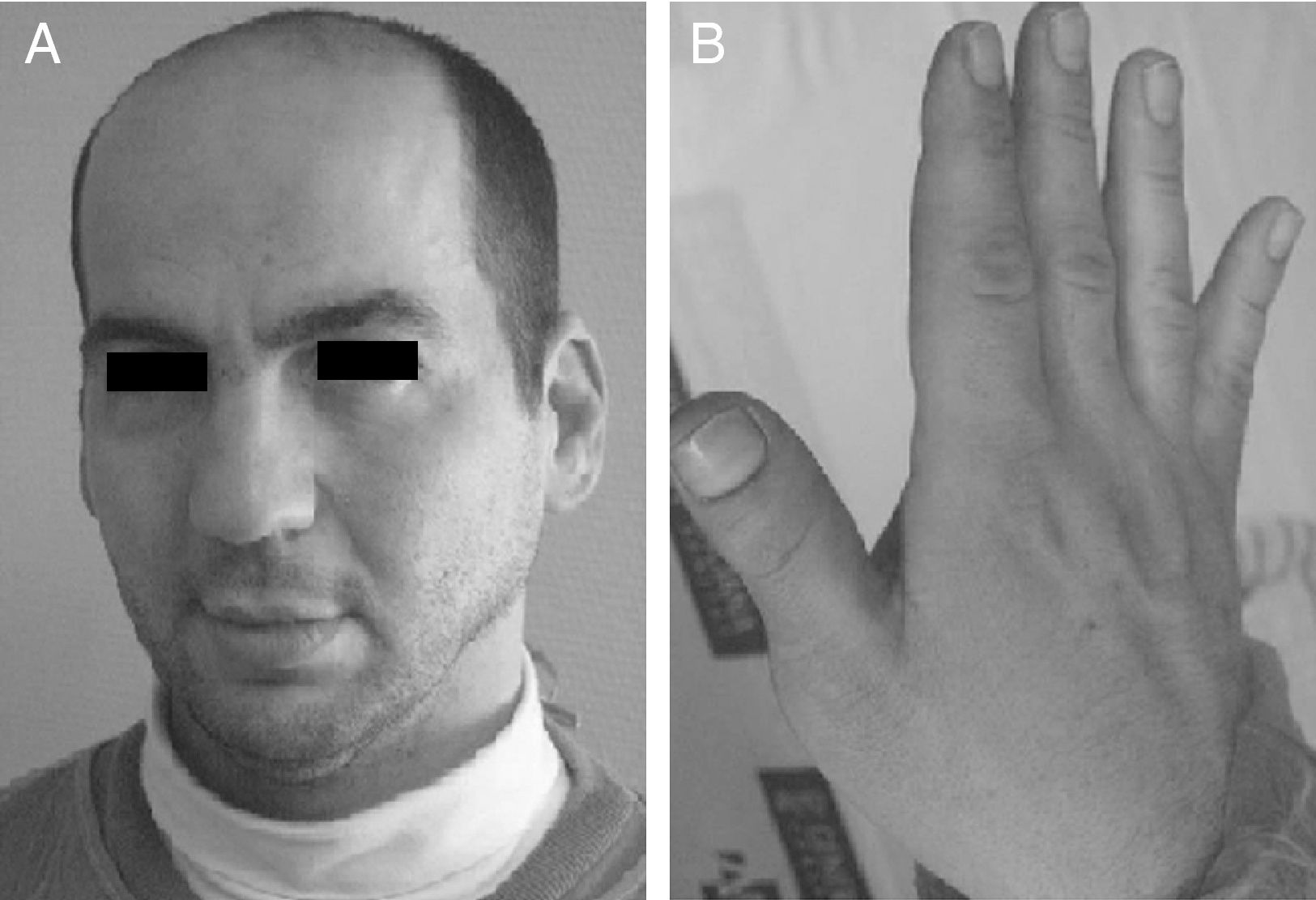
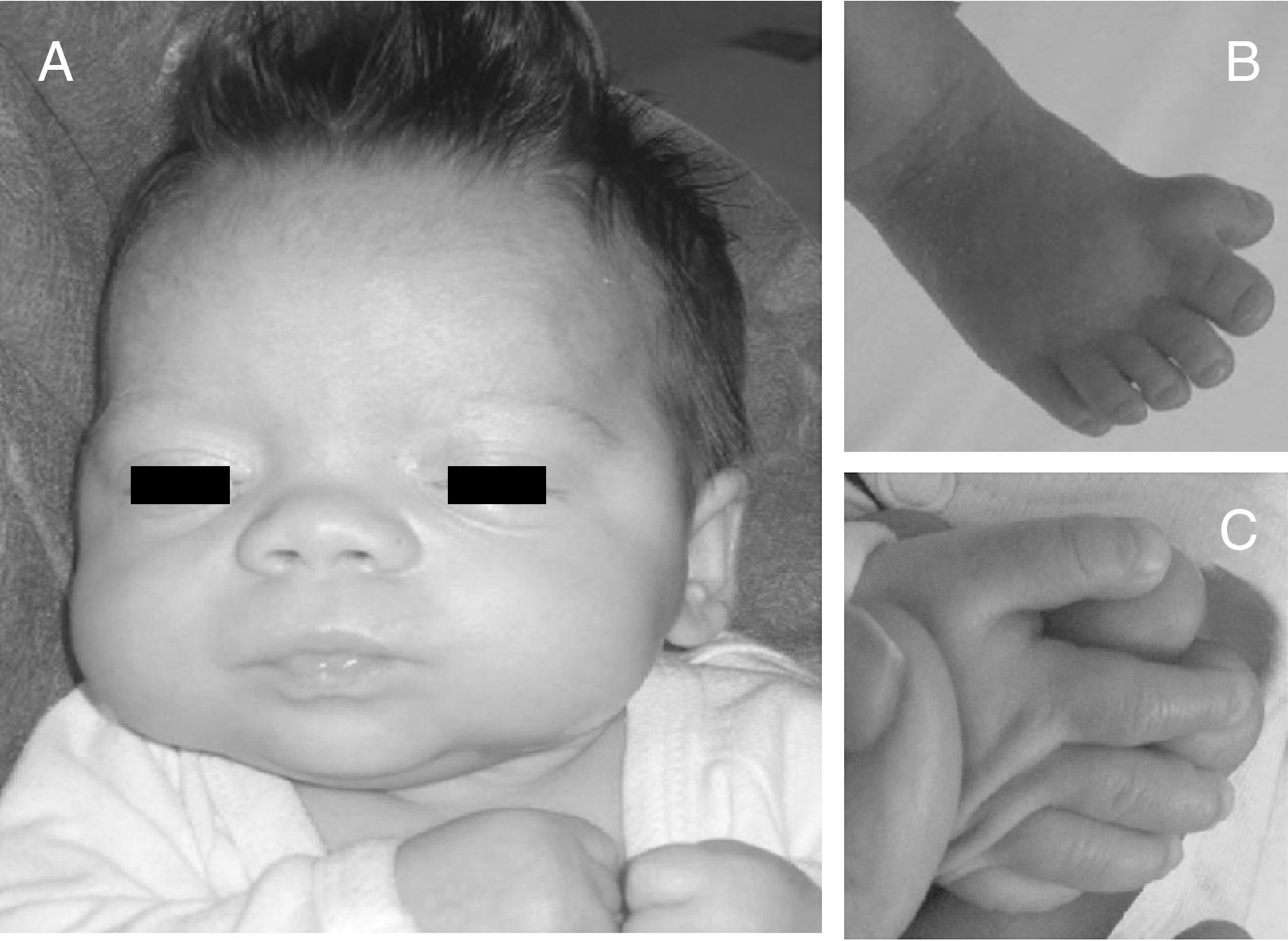
Exploración física y pruebas complementarias: peso, talla y perímetro craneal: < P5. La niña presenta polidactilia preaxial de ambos pies (6 dedos completos en ambos pies, a expensas de duplicación del primer dedo), sindactilia del 3.°-4.° de ambos pies, pulgares grandes y de implantación alta, frente amplia e hipertelorismo con raíz nasal ancha (distancias interpupilar e intercantal interna > 3 DS) (fig. 2). Hemograma y bioquímica, normales. Ecografía cerebral y abdominal, normales. Estudio cardiológico, normal.
Evolución: durante su ingreso, presentó ictericia asociada a la prematuridad. Ante la presencia de rasgos físicos compatibles, en ella y en su padre, junto con los antecedentes familiares de polidactilia, se valora la posibilidad diagnóstica de GCPS, solicitándose estudio genético a la recién nacida y a sus progenitores. Se remite a consulta de traumatología infantil para valoración y tratamiento de la polidactilia y se ofrece seguimiento madurativo en consulta externa de neonatología, tras su alta hospitalaria a los 14 días de vida.
Estudios genéticos: se realizó un estudio cromosómico convencional en sangre periférica que, con un nivel de bandeo GTG de 550, no detectó anomalías numéricas y/o estructurales. Simultáneamente, se realizó un microarray CGH Cytoarray (44K) (Agilent Technologies, Santa Clara, CA, EE. UU.), detectándose una microdeleción en la citobanda 7p14.1 de 1,5 Mb (chr7: 41410480-42923255) (NCBI Build 36.1. Sonda inicio: 0240_44224c7_1_1207_s_PSO-60-0076. Sonda fin: 0240_45807c7_1_1099_s_PSO-60-0716) (fig. 3). Esta deleción, situada por debajo del poder de resolución de la citogenética convencional, afecta a 3 genes: INHBA (codifica la proteína inhibina A), PSMA2 (codifica la subunidad 2 del proteosoma) y GLI3 (codifica un factor de transcripción que actúa tanto como activador como represor de genes de la ruta metabólica sonic hedgehog durante el desarrollo embrionario). Además, la deleción incluye un seudogén (AC073852.4) y un marco abierto de lectura (C7orf25). La misma deleción está presente en el padre de la paciente.
Discusión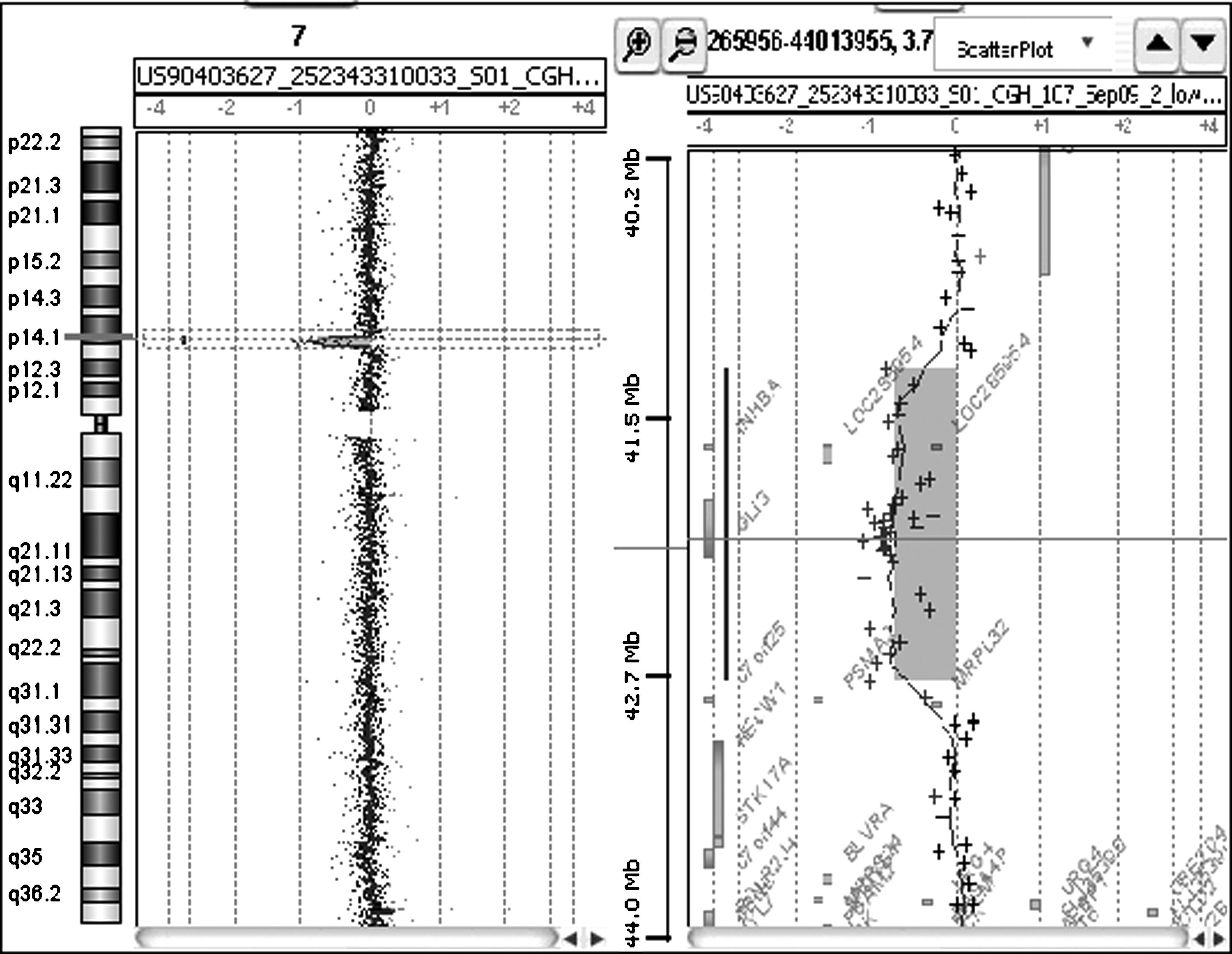
Las manifestaciones clínicas de la GCPS son poco específicas. La polidactilia, por ejemplo, puede aparecer en más de 100 patologías distintas1. Además, las manifestaciones clínicas se solapan con las que aparecen en la polidactilia preaxial tipo 4 (OMIM 174700) y los síndromes de Pallister-Hall (OMIM 146510), acrocalloso (OMIM 200990), Carpenter (OMIM 201000), Gorlin (OMIM 109400) y Teebi (OMIM 145420) (tabla 1). Por otra parte, aproximadamente en el 20% de los casos de GCPS se detecta una deleción de tamaño variable en la citobanda 7p14.1. Si esta deleción es superior a 1 Mb1, el fenotipo es más severo, y es más frecuente la patología neurológica (retraso mental, convulsiones). Además, es frecuente encontrar normo o microcefalia1, en vez de macrocefalia. Estos casos deben encuadrarse en la denominación GCPS-CGS. En el caso que describimos, la deleción (1,5 Mb) afecta a 3 genes. Uno de ellos es INHBA, que codifica una proteína (inhibina A) que dimeriza formando activina A. Este dímero tiene un efecto mitogénico sobre las células osteoblásticas8; esta actividad está regulada por la producción de folistatina por parte de los osteoblastos. Además, ratones activina A deficientes se desarrollan a término pero mueren en el periodo perinatal y presentan alteraciones dentarias y paladar hendido, lo que demuestra que la activina A puede tener un papel durante el desarrollo craneofacial9. Como hipótesis para explicar la microcefalia de nuestra paciente, pensamos que la haploinsuficiencia de INHBA (y, por tanto, la producción disminuida de activina A) podría conducir a una reducción de la actividad mitogénica osteoblástica durante el desarrollo embrionario craneofacial. No obstante, factores ambientales como el tabaquismo materno podrían tener un efecto aditivo sobre el fenotipo. En estos casos, con una sospecha diagnóstica de GCPS pero con un fenotipo más severo, debe indicarse la realización de un microarray-CGH para detectar posibles deleciones, antes que realizar un estudio mutacional por secuenciación de GLI3. En este sentido, se han diseñado arrays específicos para el síndrome con una mayor densidad de sondas en la región de GLI3, con un espaciado medio entre las sondas de 730bp (zoom-in CGH)10.
Manifestaciones clínicas de la cefalopolisindactilia de Greig y síndromes solapados
| PSD tipo 4 | GPS | GPS-CGS | PHS | ACLS | Carpenter | Gorlin | Teebi | |
|---|---|---|---|---|---|---|---|---|
| Agenesia de cuerpo calloso | ••• | |||||||
| Polisindactilia preaxial | ••• | ••• | ••• | ••• | •• | |||
| Macrocefalia | ••• | • | ••• | ••• | ||||
| Hipertelorismo | •• | •• | ••• | ••• | ||||
| Retraso mental | • | ••• | • | ••• | • | |||
| Convulsiones | •• | • | •• | |||||
| Polidactilia post-axial/central | • | •• | ••• | •• | • | • | ||
| Hamartoma hipotalámico | ••• | |||||||
| Úvula bífida | •• | |||||||
| Displasia frontonasal | ••• | |||||||
| Tumores de células basales | ••• | |||||||
| Acrocefalia | ••• |
ACLS: síndrome acrocalloso; GPS: cefalopolisindactilia de Greig; GPS-CGS: cefalopolisindactilia de Greig-síndrome de genes contiguos; PHS: síndrome de Pallister-Hall; PSD tipo 4: polisindactilia tipo 4.
Como estrategia diagnóstica genética, ante una sospecha clínica de GCPS debe indicarse como primera prueba un cariotipo convencional (bandeo GTG, nivel 550) que puede identificar anomalías estructurales aparentemente balanceadas, así como grandes deleciones (> 3 Mb). En segundo lugar, si el estudio citogenético no detecta anomalías y el cuadro clínico sugiere un síndrome de genes contiguos, debe indicarse un array-CGH. En el resto de las situaciones debe valorarse la secuenciación de GLI3.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
