


En los últimos años, el conocimiento creciente de la patogénesis de la mastocitosis ha permitido avances en el diagnóstico, el tratamiento y el pronóstico de esta enfermedad1. Los pacientes con mastocitosis y mutaciones D816V en el gen KIT pueden beneficiarse del tratamiento con midostaurina, un inhibidor de tirosina cinasa (ITC) oral multidiana2. En niños con mastocitosis son más frecuentes otras mutaciones, y estos pacientes han respondido a otros ITC como el imatinib, el nilotinib o el masitinib1. El imatinib se ha utilizado con éxito en pacientes pediátricos con mastocitosis grave refractaria a la terapia convencional1-5.
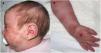
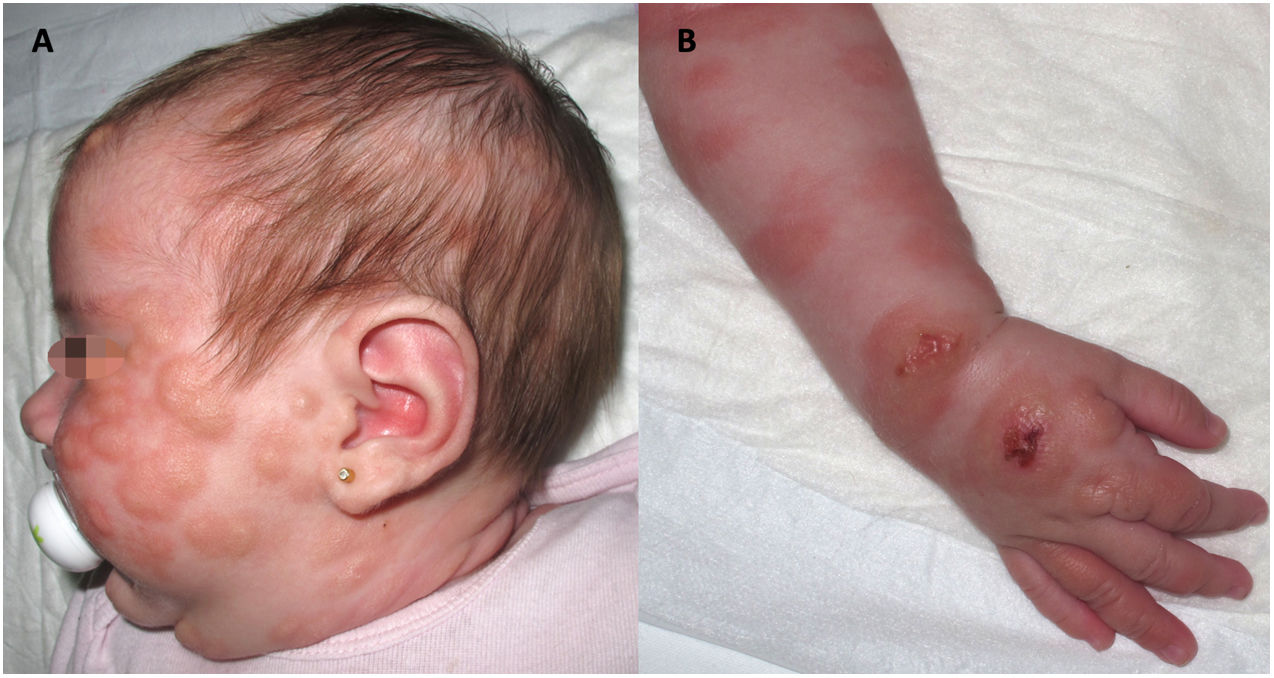
Presentamos el caso de una niña con mastocitosis cutánea difusa de inicio en el primer mes de vida, con placas extensas y lesiones ampollosas por la cara y el cuerpo (fig. 1), asociadas a eritema, dolor abdominal y diarrea. El diagnóstico se confirmó mediante biopsia cutánea. En los primeros meses de vida hubo elevación de los niveles de triptasa. La paciente mostró una respuesta limitada al tratamiento con esteroides, antihistamínicos, cromoglicato y ketotifeno. A los 4años de edad, el estado de la paciente empeoró, con desarrollo de jaquecas frecuentes y episodios de hipotensión. Fue tratada con fototerapia con psoraleno y rayos uva (PUVA) dos veces por semana, consiguiéndose con ello una mejoría inicial de los síntomas y las lesiones cutáneas, con pérdida progresiva de la efectividad a lo largo de 10meses de tratamiento.
A los 8años de edad (fig. 2A), los síntomas asociados al calor, el ejercicio y los desencadenantes emocionales empeoraron. El nivel de triptasa se mantuvo normal. Una nueva biopsia cutánea reveló un infiltrado de mastocitos con citoplasma granular c-KIT+, triptasa+, CD30+ y CD25−. Se realizó estudio de médula ósea, con recuentos y morfología celular normales. El ensayo ASOqPCR (TaqMan) no detectó la variante D816V en el gen KIT con. No obstante, se aisló un 0,0012% de mastocitos mediante la técnica de clasificación de células activadas por fluorescencia (FACS) con inmunofenotipo c-KIT+, triptasa+, CD25− y CD30−. La secuenciación Sanger de los mastocitos de médula ósea purificados mediante FACS no evidenció la presencia de la variante D816V, detectándose en cambio el polimorfismo M541L en el gen KIT. En base a este hallazgo, se inició tratamiento con imatinib a una dosis de 100mg al día, manteniendo el tratamiento con cromoglicato a una dosis de 200mg dos veces al día y ketotifeno a una dosis de 1mg dos veces al día y con adición de esteroides y antihistamínicos orales durante las crisis.
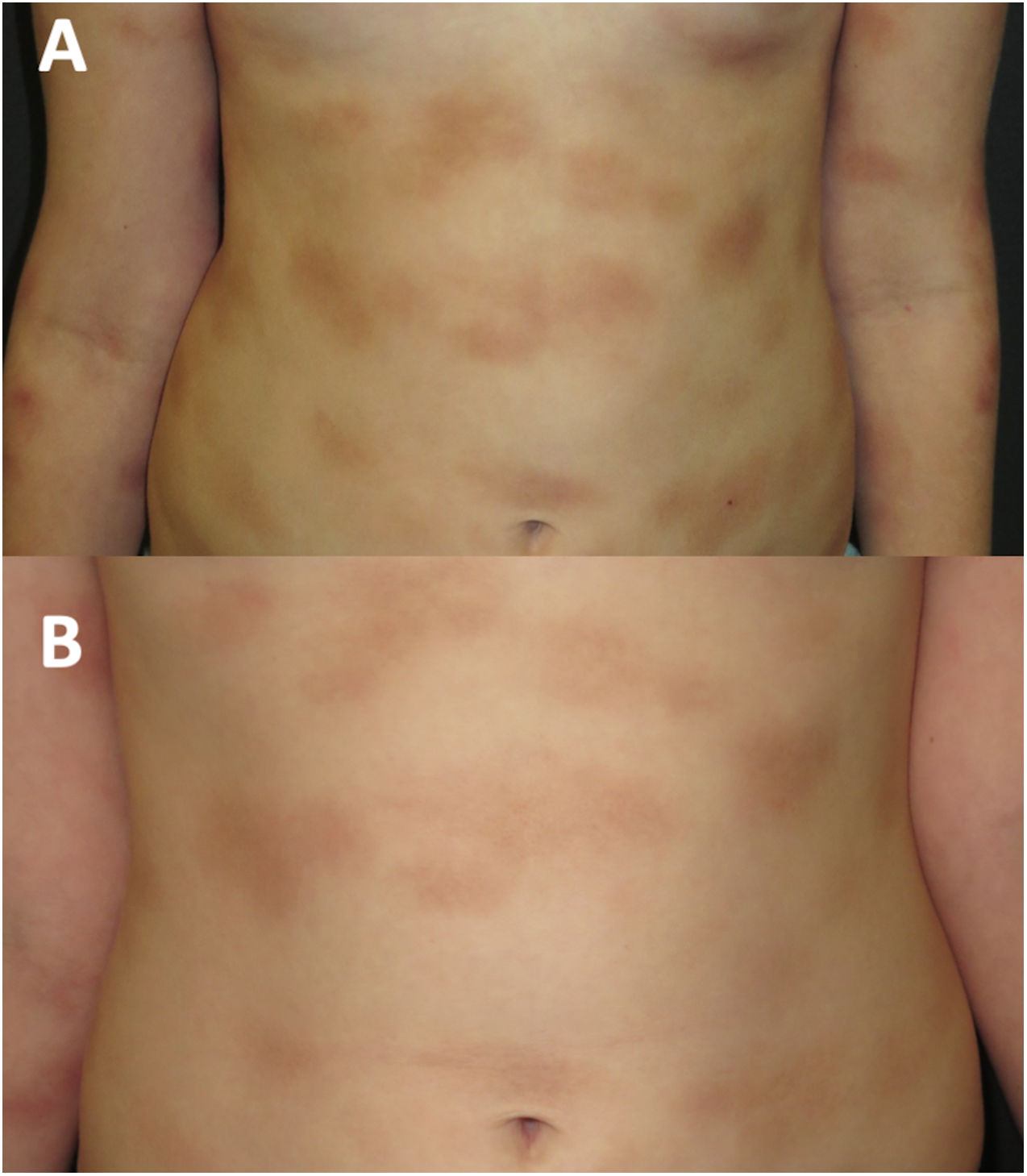
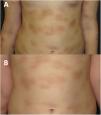
Durante los cuatro años de seguimiento, la respuesta al imatinib ha sido satisfactoria, con buena tolerancia, atenuación de la infiltración en las lesiones cutáneas (fig. 2B) y mejoría de los síntomas sistémicos. Los episodios ocasionales de mareo se han resuelto con dexclorfeniramina maleato oral. El crecimiento de la paciente ha sido normal, no se han detectado efectos adversos y las analíticas y los niveles de triptasa se han mantenido en la normalidad.
Se han descrito mutaciones activantes del receptor con actividad tirosina cinasa KIT en contexto de distintas neoplasias y en la mastocitosis cutánea difusa. El imatinib es un ITC oral autorizado para el tratamiento de la mastocitosis sistémica en pacientes con mutaciones en el gen KIT fuera del exón 17, pero no suele ser efectivo en enfermedades asociadas al polimorfismo D816V2. El polimorfismo M541L encontrado en nuestra paciente se ha asociado a la mastocitosis infantil y a una sensibilidad mayor al tratamiento con imatinib3.
Entre los efectos adversos asociados al uso de imatinib se encuentran las náuseas, los vómitos, la diarrea, la elevación de transaminasas hepáticas, la cardiomiopatía, la anemia, la trombocitopenia, la granulocitopenia, el edema, el exantema y una velocidad de crecimiento disminuida en niños4,5.
Si bien la experiencia con el uso de imatinib en niños con mastocitosis es limitada, este fármaco podría ser una alternativa en pacientes con mastocitosis cutánea difusa y síntomas graves refractarios a las terapias convencionales.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
A la familia y a la paciente, que colaboraron en la publicación de este caso y, por lo tanto, contribuyeron a avanzar en el conocimiento sobre esta enfermedad.
A los doctores Victor J. Asensio y María Carmen Vidal, del Servicio de Genética, por sus comentarios acerca del manuscrito. A la doctora Ana Bauzá por contribuir con las fotografías de la paciente en los primeros meses de vida.
