


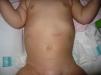

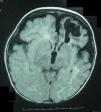
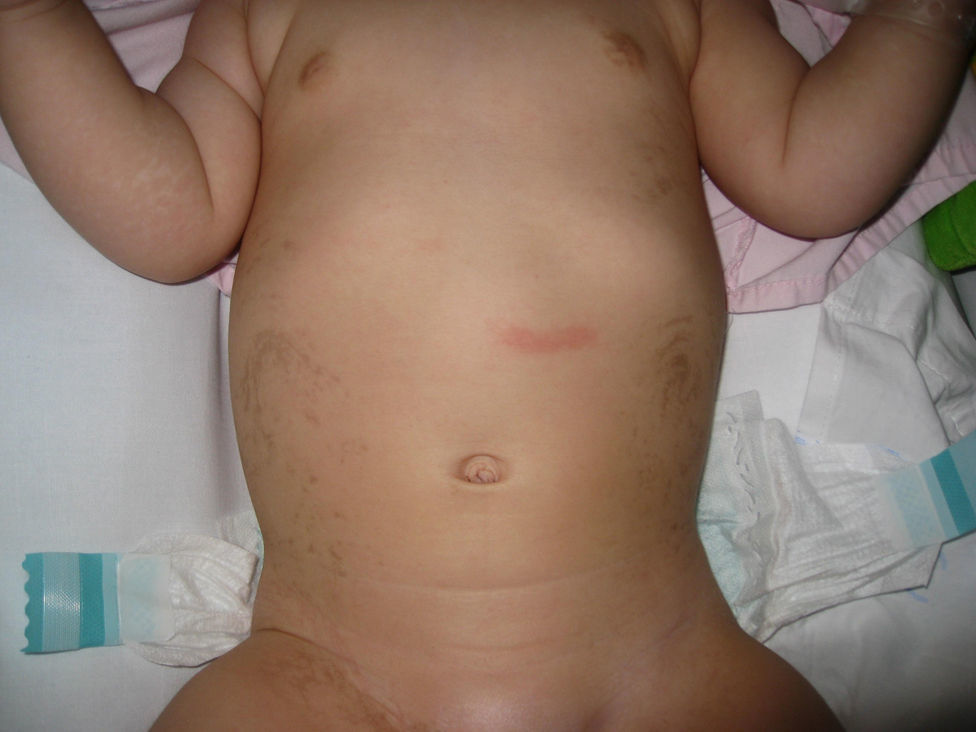
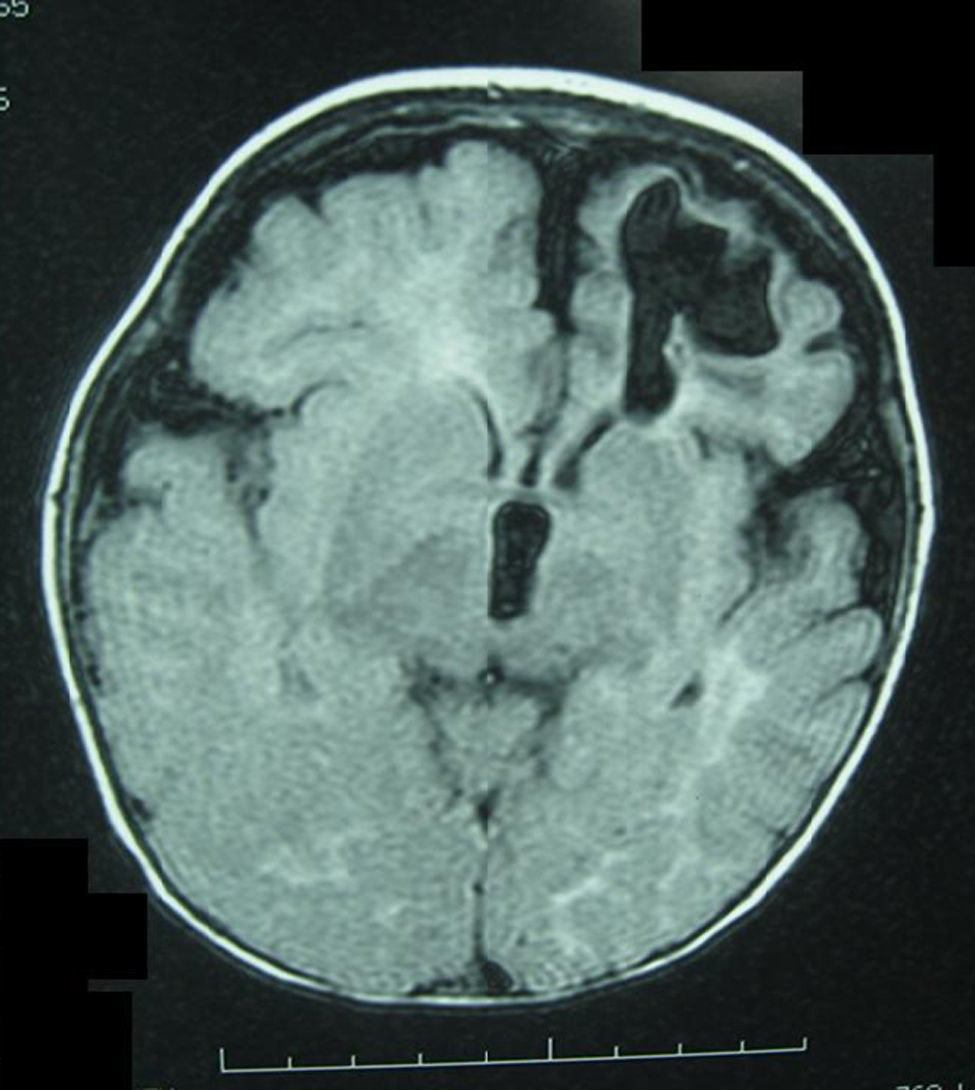
Lactante mujer de 8 meses de edad que consultó por hemiparesia derecha asociada a retraso psicomotor. Procedía de gestación y parto sin incidencdias, y no había antecedentes familiares de abortos o patologías neurológicas. En la exploración, además de la hemiparesia, se observaban lesiones hiperpigmentadas en abdomen, de disposición lineal y aspecto sinuoso-arremolinado, que seguían las líneas de Blaschko (figs. 1 y 2). Según la familia, estas lesiones estaban presentes desde el nacimiento y no habían tenido un aspecto diferente en ningún momento de la evolución. Se realizó una resonancia magnética craneal, que mostró una discreta atrofia corticosubcortical asociada a una porencefalia frontal izquierda (fig. 3). El estudio genético familiar reveló una deleción de los exones 4–10 del gen NEMO únicamente en la paciente y confirmó la sospecha clínica de incontinentia pigmenti (IP). Actualmente la niña presenta el espectro completo del síndrome, con alteraciones cutáneas, neurológicas, dentales y alopecia.

La IP es una enfermedad multisistémica rara de origen ectodérmico, secundaria a una mutación en el gen NEMO (Nuclear Factor κB Essential Modulator). El diagnóstico de sospecha es clínico y se basa en los criterios propuestos por Landy y Donnai en 1993. En su forma de presentación típica (el 92% de los casos), los pacientes presentan lesiones cutáneas que siguen una secuencia de cuatro fases. Presentamos una paciente con inicio clínico en fase 3. Esta forma de presentación es muy poco frecuente, y se ha planteado la hipótesis de que las primeras dos fases de la enfermedad ocurran de forma prenatal.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
