





El hipoaldosteronismo congénito aislado es una enfermedad infrecuente que se manifiesta como un síndrome pierde sal durante la lactancia. La deficiencia de aldosterona sintetasa debida a mutaciones en el gen CYP11B2 es la causa subyacente más frecuente. Al margen de las anomalías electrolíticas clásicas (hiponatremia e hiperkaliemia), no se han descrito hasta el momento otras patologías extra-suprarrenales. Se describe un caso de hipoaldosteronismo congénito debido a una mutación sin sentido en homocigosis (Thr318Met) en CYP11B2. Además del cuadro clínico clásico, el paciente presentó una pérdida auditiva neurosensorial no filiada.
Isolated congenital hypoaldosteronism is a rare disorder that presents as chronic salt-wasting syndrome during infancy. Aldosterone synthase deficiency due to mutations in CYP11B2 is the underlying cause in most cases. Apart from the classical electrolyte disturbances (hyponatremia and hyperkalemia), no other extra-adrenal features have been described to date. We report a male child with congenital hypoaldosteronism due to a homozygous missense mutation (Thr318Met) in CYP11B2 who also presented with unexplained sensorineural hearing loss.
Un lactante varón de 2 meses y medio de edad ingresó en el hospital por presentar escasa ganancia ponderal desde el nacimiento, que se había producido por cesárea programada debido a presentación podálica a las 38 semanas de gestación, tras un embarazo sin complicaciones. No existían antecedentes familiares de consanguinidad ni otros datos de interés. Al nacimiento no se apreciaron anomalías y, tanto el peso (3.390g), como la longitud (49cm) y el perímetro cefálico (37cm) fueron adecuados para la edad gestacional. El paciente fue alimentado inicialmente con lactancia materna exclusiva. A las 8 semanas de edad pesaba solo 3.780g por lo que suplementó la dieta con fórmula de inicio, lo cual no produjo ninguna mejoría significativa. Salvo un leve estreñimiento, el paciente no presentaba ningún otro síntoma.
Al ingreso, su peso era de 3.850g (−3DE) y su longitud de 55cm (−2DE). La tensión arterial (90/50mmHg) y el pulso (80 latidos por minuto) eran normales. La exploración física general fue normal. Los análisis de sangre evidenciaron hiponatremia e hiperkaliemia graves, por lo que se sospechó un síndrome pierde sal. El estudio de la función suprarrenal (tabla 1, parte superior) mostró niveles plasmáticos normales de cortisol, 17-hidroxiprogesterona y andrógenos suprarrenales, excluyendo los diagnósticos de hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa y de hipoplasia suprarrenal congénita. Dado que el paciente presentaba una actividad de renina plasmática muy elevada junto a una concentración inapropiadamente normal de aldosterona se planteó el diagnóstico de hipoaldosteronismo primario congénito. Los niveles de electrolitos no mejoraron tras la administración intravenosa de sodio (hasta 15mg/kg/d). En cambio, el tratamiento con 9α-fluorhidrocortisona oral (25μg/kg/d) junto a suplementos orales de cloruro sódico condujo a la normalización inmediata del estado hidroelectrolítico y el crecimiento.
Datos electrolíticos y hormonales antes y después del tratamiento
| Patiente | Rango normal | |
| En el momento del diagnóstico (sin tratamiento) | ||
| Sodio (mEq/l) | 123 | 135–145 |
| Potasio (mEq/l) | 7,4 | 3,5–5,5 |
| Cortisol (ng/ml) | 195 | 78–262 |
| 17-hidroxiprogesterona (ng/ml) | 1,92 | 0,10–1,48 |
| Δ4-androstendiona (ng/ml) | 0,29 | 0,06–0,60 |
| DHEA-S (ng/ml) | 63 | 24–360 |
| Aldosterona (ng/ml) | 1,07 | 1,0–1,15 |
| Actividad renina plasmática (ng/ml/h) | 23,6 | 0,1–1,9 |
| Tratamiento: 9α-fluorohidrocortisona | ||
| Corticosterona (ng/ml) | 11,60 | 0,29–9,37 |
| 18-hidroxicorticosterona (ng/ml) | 4,96 | 0,20–5,30 |
| Aldosterona (ng/ml) | 0,13 | 0,11–0,67 |
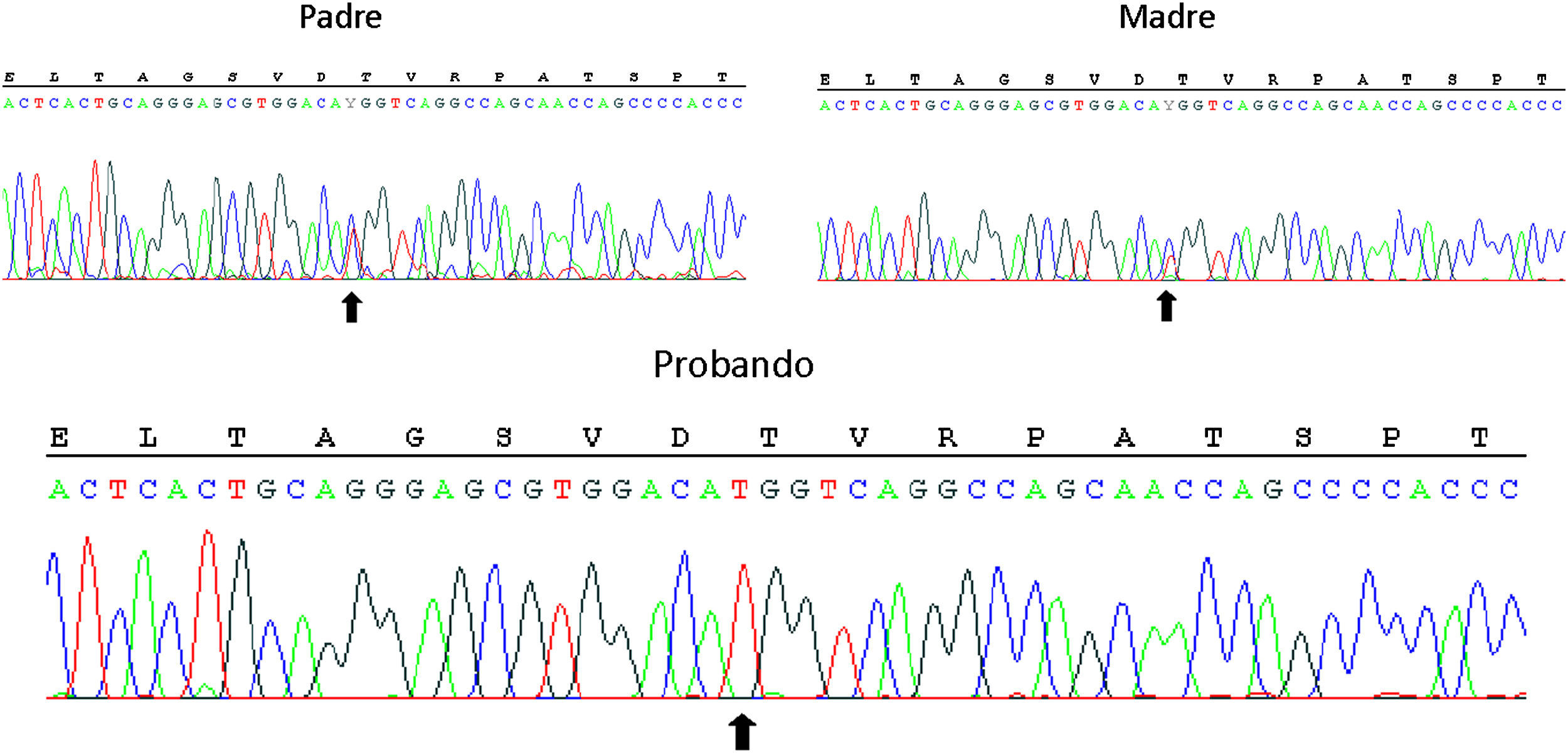
Para orientar mejor el diagnóstico, se amplió el estudio hormonal (tabla 1, parte inferior). El marcado aumento de la relación 18-hidroxicorticosterona (18-OHB)/aldosterona hasta 38,2 a pesar del tratamiento con 9α-fluorhidrodrocortisona sugirió una deficiencia de corticosterona metiloxidasa (CMO) tipo II1, que se confirmó tras el hallazgo de una mutación sin sentido en homocigosis (c.953C>T, p.Thr318Met), previamente descrita2, en el gen CYP11B2 (fig. 1). Ambos progenitores, asintomáticos, son heterocigotos para la mutación.
A la edad de 2 años se evidenció una deficiencia auditiva, que no mejoró con la colocación de drenajes transtimpánicos. La audiometría reveló una hipoacusia neurosensorial bilateral moderada, con el umbral auditivo a 60dB (fig. 2), sin anomalías estructurales en el oído interno. Desde entonces, el paciente utiliza un audífono externo.
![Audiometría efectuada a los 4 años de edad, mostrando pérdida auditiva bilateral moderada. o-o Conducción aérea derecha; x-x Conducción aérea izquierda; [Conducción ósea derecha] Conducción ósea izquierda.](https://static.elsevier.es/multimedia/16954033/0000007300000001/v1_201304301907/S1695403310002109/v1_201304301907/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w9/qVHBXBqbSQ7FNUvNof+6+l4v03CmyaR9Rm+q8TRfDEOS68CZK9bYY3KxU3zGztahdLNfZYYdbBtHnyfwstU0rc1rKxRxubw9ktHG/GVzYUK1Zk30m8YOuORaiFdOxHSFe+CzIoQkoEJkpWJ3IEcLVUpgH6+tIVR3hiFUuUso9V/+xOoSCBw+USNjpFMlzkNIstv2AiQR7QoEKlYenwS1VHA/i+rKWH6mgOWWWYYG7t1RNH1OjkK5XZorHH0eDe/TmfZ9hlvyyf2iu+xmms1kA=)
En la actualidad el paciente tiene 5 años, toma 25μg of 9α-fluorohidrocortisona al día y se encuentra bien adaptado. Su talla es acorde a la talla parental.
DiscusiónLa aldosterona, la hormona mineralocorticoide más importante, estimula la reabsorción de sodio y la secreción de potasio en los túbulos renales distales. Su ausencia congénita produce un síndrome de pérdida de sal agudo precoz tras el nacimiento. Aunque la mayoría de los casos de deficiencia de aldosterona son secundarios a deficiencia de 21-hidroxilasa, se han identificado otras causas, entre las que se encuentran la hipoplasia suprarrenal congénita, el pseudohipoaldosteronismo y el hipoaldosteronismo congénito aislado.
Este último, también conocido como deficiencia de corticosterona metiloxidasa (CMO), es una enfermedad autosómica recesiva poco frecuente causada por alteraciones en la última etapa de la biosíntesis de aldosterona, generalmente debida a la presencia de mutaciones en el gen CYP11B2 (OMIM ¿124080), que codifica la aldosterona sintetasa1. Esta enzima cataliza 3 reacciones secuenciales: la hidroxilación de la 11-deoxicorticosterona (11-DOC) a corticosterona (B); la hidroxilación de la corticosterona a 18-hidroxicorticosterona (18-OHB) y la oxidación final de la 18-hidroxicorticosterona a aldosterona.
Esta enfermedad se manifiesta en la lactancia como un síndrome pierde sal reversible, acompañado de hipoaldosteronismo hiperreninémico. Existen 2 formas bioquímicas distintas, que sin embargo son clínicamente indistinguibles. En la deficiencia de CMO tipo I (OMIM #203400), el paciente carece por completo de actividad 18-hidroxilasa, y la aldosterona es indetectable en plasma. Por el contrario, la deficiencia de CMO tipo II (OMIM #6105600) está causada por una reducción parcial aislada de la actividad de 18-oxidasa. En esta situación, los niveles séricos de aldosterona pueden ser bajos o normales, a costa de una concentración elevada de 18-OHB. Como consecuencia de ello, la deficiencia de CMO tipo II se caracteriza por un marcado incremento del cociente 18-OHB/aldosterona. El hipoaldosteronismo aislado es una enfermedad genéticamente heterogénea y algunos pacientes no presentan mutaciones en el gen CYP11B2 sino en el gen NRB01 (DAX1)3.
El paciente descrito presenta un síndrome pierde sal de presentación precoz producido por una mutación (Thr318Met) en homocigosis en el gen CYP11B2. La mutación anula la actividad de la proteína mutante2. Junto al cuadro clínico clásico, el paciente también presenta una pérdida auditiva neurosensorial bilateral de causa desconocida.
Se han identificado receptores para la aldosterona en el oído interno, especialmente en la cóclea (estría vascular y células epiteliales de la prominencia)4, donde su actividad contribuye al mantenimiento de concentraciones elevadas de potasio y bajas de sodio en la endolinfa, permitiendo así una transducción adecuada de las señales auditivas. Recientemente, se ha comunicado que la presencia de presbiacusia se asocia con niveles bajos de aldosterona en ancianos5. Por otro lado, los glucocorticoides constituyen el tratamiento habitual para varias anomalías auditivas, siendo el receptor mineralocorticoide su principal diana en el oído interno6. Todo ello sugiere que la deficiencia congénita de aldosterona podría ejercer un efecto deletéreo sobre el sistema auditivo en desarrollo. Sin embargo, hasta la fecha no se han comunicado alteraciones auditivas en los pacientes con deficiencia de aldosterona, ya sea aislada o combinada con deficiencia de otras hormonas suprarrenales, por lo que esta posibilidad es poco probable.
Por el contrario, la presencia de una mutación poco frecuente en homocigosis en el paciente pese a la ausencia de consanguinidad conocida aumenta la posibilidad de que los progenitores estén remotamente emparentados a través de un ancestro común. En este contexto, no puede excluirse que la hipoacusia sea debida a una segunda mutación recesiva en otro gen (herencia digénica), algo que no ha podido ser explorado en este caso dado el elevado número de genes potencialmente implicados7.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos a Felix Riepe (Universidad de Kiel, Germany) por realizar la determinación de corticosterona y 18-hidroxicorticosterona y la secuenciación del gen CYP11B2. Asimismo, agradecemos al Instituto de Salud Carlos III su apoyo al CIBERobn y a la Fundación Endocrinología y Nutrición. ORC recibió una «Ayuda para contratos post-Formación Sanitaria Especializada» del «Instituto de Salud Carlos III» (FIS CM06/00013).
