






Introducción
El término hiperinsulinismo congénito engloba un grupo heterogéneo de trastornos genéticos caracterizados por una alteración en la regulación de la secreción insulínica que implica episodios recurrentes de hipoglucemia 1,2. Constituye la causa más frecuente de hipoglucemia persistente en la infancia temprana 1,3 y se calcula una incidencia global de 1 por cada 30.000-50.000 nacidos vivos 4,5. En poblaciones de tradición endogámica llega al 1 por 2.500 5,6.
El considerable avance sucedido en la última década en el terreno de la genética molecular ha contribuido a una mejor comprensión de la fisiopatología de este proceso, de modo que, hasta el momento, se conocen 5 alteraciones genéticas diferentes y la historia natural del tipo de hiperinsulinismo congénito que genera cada una de ellas. Éstas afectan a los genes de las enzimas glutamato deshidrogenasa (GDH), glucocinasa (GK) y la recientemente descubierta L-3-hidroxiacil-CoA deshidrogenasa de cadena corta (SCHAD), así como a los genes del canal de K+ ATP-dependiente de la célula β, un complejo octamérico compuesto de 2 proteínas distintas: el receptor sulfonilurea 1 (SUR1) y la subunidad KIR6.2 1,2,7-13. El daño estructural de las 3 últimas proteínas (SUR1, KIR6.2 y SCHAD) y la hiperfunción de las dos primeras (GDH y GK) determinan un estado de despolarización permanente de la célula β y, consecuentemente, una secreción insulínica continua que no responde a la concentración de glucemia 7,8,10,12,13. Hay que decir, sin embargo, que mutaciones de estos genes se han encontrado en torno al 50 % de los casos 2,4,8-10,14 y que, por tanto, es posible que haya otros genes implicados en la génesis de esta enfermedad 15. Debe añadirse también que las formas graves de inicio neonatal suelen corresponderse con las mutaciones del canal de K+ o, menos frecuentemente, de la enzima SCHAD, mientras que las formas más tardías responden a las mutaciones de las enzimas GDH, GK y, también, SCHAD 1-4,7-10,12,15.
Anatomopatológicamente, los términos "nesiodioblastosis" y "nesiodiodisplasia", tan utilizados como sinónimos de hiperinsulinismo congénito y que describían la existencia de células β aisladas o agrupadas en pequeños racimos alrededor de los conductos pancreáticos 16,17, han sido declarados como no válidos por muchos autores para designar esta enfermedad, ya que tales descripciones se han descubierto en controles normales 18 y, por otra parte, otras alteraciones histopatológicas como la hiperplasia de islotes, la hipertrofia de células β, el agrandamiento nuclear de células β o la presencia de células gigantes se han encontrado también en pacientes con hiperinsulinismo congénito 18-21. Lo que todavía prevalece es la distinción entre forma difusa y forma focal, irreconocibles clínicamente pero distinguibles desde el punto de vista anatomopatológico y molecular, ya que ambas están producidas por eventos moleculares distintos. Así pues, mientras que la forma difusa depende de una alteración de la línea germinal que afecta a todas la células del organismo y, por tanto, a todo el páncreas, la forma focal es debida a una alteración genética heredada del alelo paterno SUR1 o KIR6.2 asociada a la pérdida espontánea de material genético materno de la zona 11p15.1 restringida a un foco de células b del páncreas 1,2,7,19,22-25.
En lo que respecta al diagnóstico, los episodios de hipoglucemia deben sospecharse a edades precoces en presencia de signos y síntomas inespecíficos. En tales casos, y durante el período neonatal precoz, deben descartarse entidades como el hijo de madre diabética, el síndrome de Beckwith-Wiedeman, la asfixia perinatal o la isoinmunización Rh, aunque la recurrencia de los episodios de hipoglucemia debe poner en alerta sobre la posibilidad de un hiperinsulinismo congénito 8,26. Un segundo paso exige confirmar esta posibilidad, buscando las características bioquímicas que reflejen la acción metabólica de la insulina, esto es, el cumplimiento de los siguientes criterios: glucemia menor de 40 mg/dl con un índice insulina/glucosa igual o superior a 0,3, ácido β -OH-butírico menor de 1 mmol/l y la exclusión de otras causas metabólicas y hormonales 2,8. Es frecuente, sin embargo, tener que realizar varias determinaciones en momentos diferentes para demostrar la existencia de un estado de hiperinsulinismo y, en caso de persistir la duda diagnóstica, optar por otros parámetros analíticos como el descenso de factor de crecimiento insulinoide (IGF-BP1), una respuesta positiva al glucagón durante la hipoglucemia o una respuesta positiva a la octreótida que permita una reducción de los aportes de glucosa 8.
No obstante, cualquiera que sea el tipo de hiperinsulinismo congénito y dado el riesgo nada desdeñable de daño neurológico permanente, la prioridad en tales casos es la de iniciar un tratamiento que mantenga la normoglucemia lo más precoz y eficazmente posible. En este sentido cabe decir, sin embargo, que el avance en el campo de la terapia médica no ha sido tan satisfactoria, y la pancreatectomía es todavía el tratamiento más eficaz en los casos graves.
El objetivo de este trabajo es describir la evolución de 22 niños con hiperinsulinismo congénito seguidos en el Servicio de Endocrinología Pediátrica del hospital infantil La Paz.
Pacientes y métodos
Durante un período de 22 años, desde 1982 a 2004, hemos seguido un total de 22 pacientes con hiperinsulinismo congénito, 12 niños y 10 niñas. El diagnóstico de hiperinsulinismo permanente se estableció tras la exclusión de formas transitorias (hijo de madre diabética, síndrome de Beckwith-Wiedeman, etc.) y en presencia de episodios recidivantes de hipoglucemia con concentraciones elevadas de insulina (relación insulina/glucosa ≥ 0,3) asociados a valores disminuidos de β-OH-butirato y en ausencia de metabolopatías y déficit hormonales (GH, hormona adrenocorticotropa [ACTH] y cortisol). La determinación de amonio en ausencia de hepatopatía nos hacía sospechar la variante de hiperinsulinismo congénito debida a una alteración genética de la GDH.
El protocolo terapéutico y posterior seguimiento incluyeron:
1.Aportes de glucosa intravenosa según necesidades, asociada a alimentación frecuente (cada 2-3 h) y/o nutrición enteral nocturna a débito continuo (NEDC).
2.Administración de diazóxido, en dosis inicial de 5 mg/kg/día y máxima de 25 mg/kg/día, dividido en 3-4 dosis por vía oral; la dosis fue modificada según la respuesta de la glucemia. Cuando la dosis necesaria rebasaba los 15 mg/kg/día se asoció hidroclorotiazida a 1-2 mg/kg/día con objeto de evitar la retención hidrosalina, tan frecuentemente descrita como efecto secundario de las altas dosis de diazóxido.
3.Administración de octreótida como alternativa terapéutica en casos de mala respuesta o intolerancia al diazóxido. La dosis inicial fue de 5 μg/kg/día vía subcutánea cada 6-8 h, hasta un máximo de 40 μg/kg/día.
4.En caso de fracaso del tratamiento médico se procedió a la pancreatectomía subtotal (extirpación del 85-95 % del páncreas). La recidiva de las hipoglucemias tras la cirugía obligaba a un nuevo intento con diazóxido y/u octreótida y, ante una nueva falta de respuesta, pancreatectomía total (99 % de páncreas).
5.Evaluación clínica del crecimiento por antropometría y edad ósea. La edad ósea se estimó mediante una radiografía de la mano izquierda y se evaluó mediante el método TW2, variante mano total. El valor obtenido se situó en el canal percentilar correspondiente según los patrones de referencia para la población española (Hernández, 1987).
6.Evaluación neurológica mediante un análisis de los hitos del desarrollo psicomotor (test de Denver) e intelectual (grado de escolarización). La evaluación evolutiva por parte de neurólogos pediatras y psicólogos se llevaba a cabo cuando se detectaba cualquier anomalía en la evaluación o había antecedente de convulsión.
7.Evaluación del metabolismo de hidratos de carbono mediante la determinación anual de péptido C, hemoglobina glucosilada (HbA1C) e insulina/glucosa a través de un perfil (basal y tras desayuno) o una sobrecarga oral de glucosa si existía sospecha de intolerancia (a los 0, 30, 60 y 120 min).
8.Evaluación de la función pancreática exocrina en caso de pancreatectomía mediante Van de Kamer y quimiotripsina fecal anual.
9.Estudio molecular mediante secuenciación cíclica, y previa amplificación con reacción en cadena de la polimerasa (PCR), de los 39 exones del gen ABCC8 (SUR1) y del único exón del gen KCNJ11 (KIR 6.2), y de sus zonas de corte y empalme. Este análisis molecular se ha podido realizar en cinco de nuestros últimos pacientes diagnosticados de hiperinsulinismo congénito.
Resultados
De los 22 afectados de hiperinsulinismo congénito, 15 (73 %) tuvieron un peso adecuado para la edad gestacional y 6 (27 %) fueron grandes para la edad gestacional (> 2 desviación estándar [DE]) (tabla 1). La hipoglucemia fue documentada en la primera semana de vida en 17 casos (77 %) y el resto (23 %) a una edad media de 5,1 meses (45 días-13 meses). Los síntomas iniciales más frecuentes fueron: hipotonía, 54 %; convulsiones, 45 %; cianosis, 31 %, y, en menor porcentaje, otros síntomas como sudoración, palidez, apnea o hipotermia. Entre los antecedentes familiares no había nada reseñable desde el punto de vista endocrinológico.
Todos los niños recibieron aportes de glucosa intravenosa, a una media de 16 mg/kg/min (13-28 mg/kg/min), además de alimentación frecuente (cada 2-3 h, con o sin sonda nasogástrica) y, en algunos casos, NEDC nocturna.
La administración de diazóxido fue necesaria también en todos los casos. La dosis inicial (5-10 mg/kg/día) se administró entre 2 días y 3,6 meses después de iniciados los síntomas (media de 32,1 días). En 5 casos (22 %) se utilizó como única terapia, de los cuales dos continúan con ella y a tres se les ha suspendido con 11 meses, 12 meses y 3 años de edad, respectivamente. Los efectos secundarios observados fueron la hipertricosis en tres y la retención de líquidos en dos
La octreótida, empleada de forma sistemática en nuestro centro a partir del año 1994, quedó reservada para aquellos pacientes que no respondieron a diazóxido (8 de 12, el 66 %). Todos ellos presentaron, pese a una buena respuesta inicial, un fenómeno de taquifilaxia que obligó a un incremento de la dosis y, finalmente, a la pancreatectomía. El caso 19, por preferencia de los padres a la terapia médica, ha presentado buena evolución con dosis elevadas de octreótida como único tratamiento; éste se ha logrado retirar en el último año.
La pancreatectomía subtotal acabó realizándose en 16 pacientes (72 %) por falta de respuesta al tratamiento médico. La primera pancreatectomía se realizó a una edad mínima de 13 días y máxima de 4 años. Después de la primera pancreatectomía, siete evolucionaron favorablemente, cinco precisaron de diazóxido, tres diazóxido más octreótida y uno falleció. De los ocho que precisaron terapia médica tras la cirugía, uno evolucionó favorablemente al poder suspenderse el diazóxido con 4,5 años de edad; el resto, sin embargo, tuvieron que ser sometidos a una segunda pancreatectomía. En este grupo de pancreatectomizados por segunda vez, cuatro han evolucionado satisfactoriamente (tres llegaron a precisar insulinoterapia transitoriamente), dos han requerido diazóxido (a uno se le suspendió con 6 años 8 meses y el otro continúa con una dosis de 4,4 mg/kg/día) y uno ha llegado a precisar una tercera cirugía tras intento frustrado con terapia médica combinada (diazóxido y octreótida) (fig. 1).
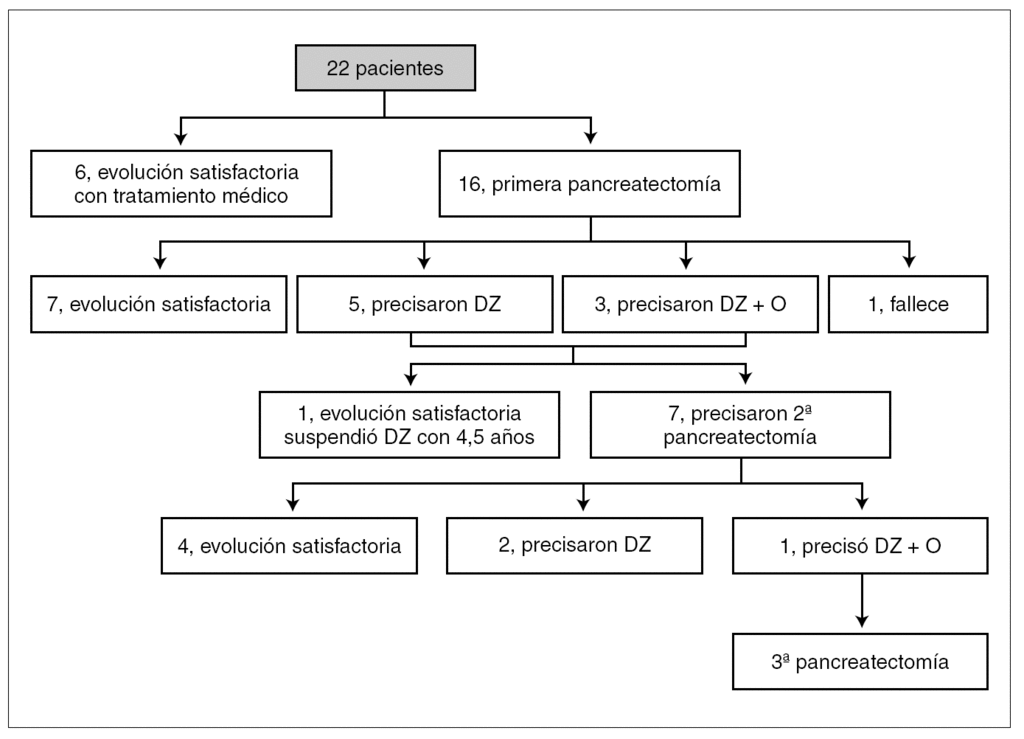
Figura 1. Algoritmo terapéutico evolutivo de los 22 pacientes con hiperinsulinismo congénito. DZ: diazóxido; O: octreótida.
La anatomía patológica en todos los casos operados fue informada como nesidioblastosis (hiperplasia de células β en racimos alrededor de conductos pancreáticos), y sólo en los cuatro últimos se ha constatado el grado de extensión de la lesión (difusa), por lo que no se ha podido obtener una proporción fiable de formas difusas frente a focales.
El estudio genético se ha realizado recientemente en algunos pacientes que presentaron hiperinsulinismo congénito grave. Hasta el momento disponemos de cinco resultados; todos ellos presentan alteración del gen de la subunidad SUR1.
La evolución de los datos antropométricos referidos a talla y peso así, como el desarrollo puberal en los de edad más avanzada han sido normales.
Las secuelas neurológicas se han encontrado en el 28 % de los pacientes. En 5 niños (22 %) se han observado algunas alteraciones como retraso motor fino, déficit de atención, hiperactividad, retraso escolar, retraso del lenguaje y un caso, el de diagnóstico más tardío, de retraso psicomotor moderado. Sólo 3 niños precisan tratamiento anticonvulsivo en el momento actual (13 %).
Desarrollaron alteración de la tolerancia a la glucosa y/o alteración de la glucosa en ayunas 4 niños (26 %) de los pancreatectomizados, de los cuales tres han evolucionado diabetes mellitus (uno a los 7,5 años de edad tras dos pancreatectomías, otro a los 4,5 años después de tres intervenciones y otro a los 9,5 tras una cirugía). En lo que respecta a la función pancreática exocrina de los pancreatectomizados por segunda o tercera vez, seis (37 %) presentan datos analíticos de malabsorción y precisan de terapia sustitutiva.
Discusión
El término hiperinsulinismo congénito, también denominado hipoglucemia hiperinsulínica persistente de la infancia, engloba un grupo heterogéneo de trastornos genéticos caracterizados por una alteración en la regulación de la secreción insulínica que acarrea episodios recurrentes de hipoglucemia 1,2. Son cinco, hasta el momento, las alteraciones genéticas conocidas causantes de un hiperinsulinismo congénito (tabla 2 y fig. 2) y, dependiendo de donde éstas acontezcan, línea germinal o somática, la afectación será difusa o focal, respectivamente 1,2,7,19,22-25. La tabla 3 resume los defectos moleculares posibles y su relación con la extensión la lesión 8. En este sentido, distintas series revelan el 40-50 % de hiperinsulinismo congénito focales que suelen ser de una única región del páncreas 8,27, hallazgo que no puede compararse con la nuestra dado que no disponíamos del estudio anatomopatológico de las piezas más antiguas.
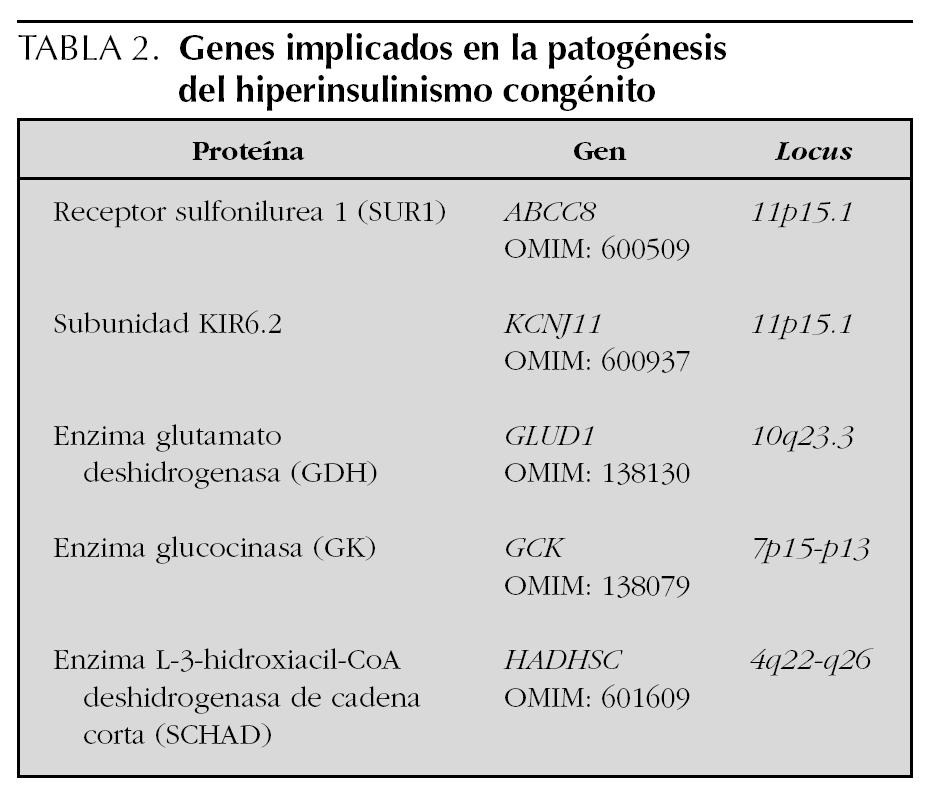
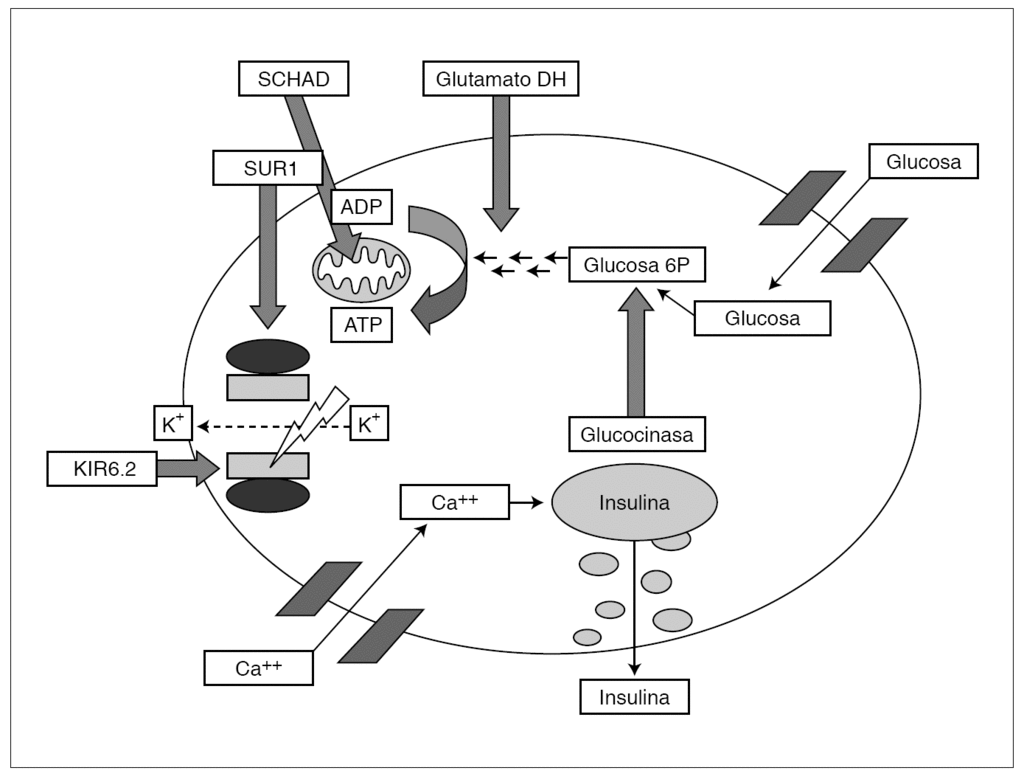
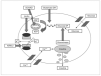
Figura 2. Secuencia funcional de la célula β y posibles alteraciones genéticas.

Clínicamente, los síntomas de hipoglucemia varían según el período de la vida en que aparezcan, y son tanto más inespecíficos cuanto menor sea la edad. El inicio neonatal es más frecuente, pues cerca del 60 % de los pacientes presentan síntomas antes de las 72 h de vida 2. Las manifestaciones en el período neonatal son convulsiones en aproximadamente el 50 % de los casos, síntomas no específicos (cianosis, palidez e hipotonía) en el 30 %, e hipoglucemia asintomática en el 20 %. Tienen poca tolerancia a períodos cortos de ayuno, precisan mayores aportes de glucosa intravenosa 1,2 y, en general, tienen mala respuesta al tratamiento farmacológico dado que predominan las mutaciones que afectan al canal de K+ (SUR1 o KIR6.2) 2,4,8. En nuestra serie presentaron síntomas en la primera semana de vida el 77 % de los niños (63 % en las primeras 72 h), con sintomatología similar a la descrita en la literatura médica. También coincide el hecho de presentar sintomatología precoz y grave los 5 casos con alteración del SUR1. Recientemente, sin embargo, Henwood et al 28 han demostrado la existencia de mutaciones menores del canal de K+ que afectarían parcialmente a la funcionalidad del mismo y explicaría los casos menos graves y de respuesta favorable al diazóxido.
Aproximadamente el 35 % de los pacientes presentan síntomas después del primer mes de vida y sólo el 5 % desarrolla síntomas después del primer año 2. En nuestra revisión, 5 casos han presentado clínica a partir del primer mes (22 %) y un caso a partir del primer año de vida (4 %). Este ligero predominio de la incidencia en la época neonatal de nuestra casuística podría deberse a que, durante muchos años, nuestro hospital ha sido referencia nacional de patología neonatal grave.
Los síntomas de presentación a estas edades son convulsiones en el 70 % de los casos o equivalente convulsivo (pérdida de conciencia, ausencias o tics) en el 20 %, y signos inespecíficos tales como temblores, irritabilidad, palidez o alteraciones de conducta en el 10 % 2,8. La sintomatología predominante de nuestros casos fueron las convulsiones. Según distintos autores, el comienzo de los síntomas en este período de la vida se ha relacionado con la hiperactividad de la GK o, más frecuentemente, de la GDH 1,2,8. También se han descrito algunos casos tardíos con defectos del canal de potasio y, recientemente, algún otro debido a alteración de la SCHAD 1,2,12.
Mención especial merece el hiperinsulinismo que deriva de la hiperfunción de la enzima GDH, también denominado síndrome de hiperinsulinismo-hiperamoniemia (HI-HA), ya que suele tener un curso clínico leve y de inicio tardío, en el cual la hipoglucemia suele ser pospandrial, después de una comida rica en proteínas, y frecuentemente se asocia hiperamonemia asintomática en grado variable. La histología suele ser difusa y la respuesta al tratamiento con diazóxido es buena 2,29,30. En nuestra revisión presentamos 2 casos de HI-HA (casos 9 y 17), aún pendientes de confirmar genéticamente, con excelente respuesta terapéutica.
Otro dato que merece ser comentado en estos pacientes es la macrosomía, ya que, aunque se ha descrito como un hallazgo característico en neonatos que cursan hiperinsulinismo, sólo unos pocos recién nacidos con hiperinsulinismo congénito son macrosómicos 8,31. Este hecho coincide con nuestra casuística, en la que sólo el 27 % tenían un peso elevado para la edad gestacional. También coincide, con excepción de 2 casos, el hecho de una mayor agresividad de la enfermedad con mayor tasa de pancreatectomía en los pacientes con hiperinsulinismo congénito y con macrosomía 1.
Para establecer el diagnóstico de hipoglucemia de origen hiperinsulinémico deben cumplirse los criterios anteriormente mencionados y, en caso de persistir, hay que sospechar hiperinsulinismo congénito. En tal situación, la identificación preoperatoria de formas focales de hiperinsulinismo congénito resulta imperativa, ya que su manejo posterior así como su pronóstico son sustancialmente diferentes a los de la forma difusa 15,32-36. Para tal efecto, se han desarrollado técnicas que permiten diferenciarlas, a saber, la determinación seriada de insulina y glucosa mediante canalización venosa pancreática vía porta y tras estimulación con calcio intraarterial 32,33 y, recientemente, la tomografía de emisión de positrones con F-fluoro- L-dopa, que ha demostrado tener claras ventajas sobre la anterior 34. Técnicas más sencillas como la respuesta insulínica periférica a la estimulación con calcio o tolbutamida no han resultado, sin embargo, eficaces en tal propósito 35,36. En nuestro hospital realizamos estos procedimientos en el caso 19 (canalización venosa pancreática a través de la vena porta) y el caso 21 (test de estimulación periférica con calcio) pero no se obtuvieron datos concluyentes en ninguno de los dos. Las técnicas de imagen convencionales (ecografía, tomografía axial y resonancia magnética) no aportan datos en este sentido ni en el diagnóstico de hiperinsulinismo congénito 2,9,37,38.
El objetivo primordial del tratamiento es mantener la normoglucemia para prevenir el daño neurológico 2,38,39. En los pacientes con hipoglucemias graves durante el período neonatal los requerimientos de glucosa pueden llegar hasta 25-30 mg/kg/día, distribuidos en forma de alimentación frecuente y fluidoterapia intravenosa de glucosa. A la vez, deben emplearse fármacos como el glucagón, la somatostatina, el diazóxido, la octreótida o el nifedipino (tabla 4).

El glucagón y la somatostatina en infusión continua pueden ser de utilidad temporal hasta establecerse el diagnóstico definitivo 2. A largo plazo, el fármaco de elección es el diazóxido por vía oral que, acoplado al receptor SUR1, permite la apertura del canal de K+ y el mantenimiento de la glucemia, si bien, la mayor parte de los casos graves neonatales no responden al mismo por requerirse de la integridad de dicho canal. El nifedipino constituye una alternativa oral poco empleada dado que se dispone de menor experiencia. Como terapia de rescate se emplea la octreótida por vía subcutánea aunque, debido a su eficacia transitoria, que responde a un fenómeno de taquifilaxia, suele emplearse mientras se procede a investigaciones que diferencien el tipo de hiperinsulinismo congénito y previo a la pancreatectomía 2,8,38,40,41.
Todos nuestros pacientes habían recibido inicialmente tratamiento médico con diazóxido, aunque sólo respondieron satisfactoriamente al mismo 5 niños, tres de ellos de presentación tardía. Esto significa una respuesta eficaz a diazóxido en 2 de 17 casos precoces (11 %) y en 3 de 5 casos tardíos (60 %), porcentajes similares a los de otras casuísticas 1,9,31. Aún en estos casos, algunos autores aconsejan la realización de técnicas que permitan diferenciar entre una forma focal y una difusa y, en caso de tratarse de una variante focal, proceder a la intervención quirúrgica pertinente 42.
La octreótida se ha utilizado de manera preoperatoria en ocho de nuestros casos (66 %) y como única terapia, por petición de los padres, en un caso de presentación neonatal grave sin respuesta al diazóxido; su evolución ha sido satisfactoria.
Cuando la terapia médica no permite un control seguro de las glucemias, el siguiente paso es la cirugía pancreática. Las formas difusas requieren de una pancreatectomía subtotal mientras que las focales podrían beneficiarse de una pancreatectomía selectiva que minimizaría el riesgo de diabetes mellitus tras la misma 2,19,27,38,43,44. En las formas difusas, sin embargo, no es infrecuente que la pancreatectomía resulte insuficiente y precise continuar con el tratamiento médico (5-25 % de los casos) o, incluso, obligue a una segunda intervención en la cual se extirpe el 99 % del páncreas. Por este motivo, la pancreatectomía subtotal inicial recomendada debe ser del 95 % 38,45-50.
Hasta ahora se ha recalcado la importancia de un diagnóstico precoz y un tratamiento eficaz en aras de evitar el daño cerebral que con frecuencia se ve en los pacientes con hiperinsulinismo congénito en forma de retraso psicomotor y/o epilepsia. Se ha comprobado una mayor tasa de tales complicaciones en aquellos casos que comenzaron durante la primera semana de vida y no respondieron adecuadamente al tratamiento médico 1,8,39,49,51-53. El porcentaje de retraso psicomotor encontrado en las distintas series es variable, desde el 0 51 al 44 %, aunque la cifra media se sitúa en torno al 30-40 % 1,31,39, algo superior comparada con la de nuestra casuística (22 %). Esta proporción asciende, según Rother et al 53, al 50 % en los niños que precisaron de pancreatectomía; en nuestra serie, sin contar los casos más recientes por su corta edad o fallecimiento, hallamos 6 de los 15 pancreatectomizados (»40 %).
En lo que respecta a la alteración del metabolismo hidrocarbonado que se observa en algunos de estos pacientes, aproximadamente el 30 % con pancreatectomía subtotal desarrollan diabetes mellitus años después de la remisión clínica, a menudo durante la pubertad; esta cifra alcanza el 70 % en los casos reoperados. Su comportamiento se asemeja a la diabetes mellitus tipo 2 ya que pueden ser tratados exclusivamente con modificaciones en la dieta, aunque casi todos acaban precisando de insulinoterapia 1,48,51. En nuestra serie, 3 pacientes de los pancreatectomizados han desarrollado diabetes mellitus a edades más tempranas que las referidas en la literatura especializada (9,1, 4,5 y 7,5 años), todos ellos con buen control y bajas necesidades de insulina. Este hallazgo puede explicarse por el hecho de que todos ellos precisaron más de una pancreatectomía y, como comentan algunos autores, porque el riesgo para desarrollar diabetes mellitus en las formas difusas no sólo es debido a la amplia resección pancreática que requieren sino también a la evolución natural de las células β hacia una pérdida de función por un posible fenómeno de apoptosis derivado del defecto genético subyacente 8,54,55. Esto último explicaría también la evolución de otros de nuestros pacientes que precisaron de medicación y que, posteriormente, acabó retirándose.
Otra complicación de la pancreatectomía es la insuficiencia pancreática exocrina. Se ha demostrado, al realizar estudios de secreción pancreática, que todos presentan una insuficiencia exocrina subclínica, sin embargo, sólo el 10 % de los niños con pancreatectomía subtotal necesitan suplementos enzimáticos 48,49. Nuestra casuística muestra 6 casos de insuficiencia pancreática exocrina, lo que representa el 37 % del total de los pancreatectomizados. La explicación de esta cifra dispar responde a lo descrito anteriormente, pues cinco de ellos habían sido operados por segunda o tercera vez; recordemos que la muestra de nuestra casuística no resulta representativa si se tiene en cuenta que también nuestro hospital es referencia quirúrgica de los casos más graves.
Finalmente, con respecto a la evolución del crecimiento, no hay estudios fiables de seguimiento a largo plazo de pacientes con tratamiento farmacológico y/o quirúrgico, aunque en series pequeñas de niños tratados con octreótida se ha observado un crecimiento normal, aun con concentraciones de IGF-I bajas 1,41; en los casos pancreatectomizados puede haber una desaceleración del crecimiento en el primer año posquirúrgico, probablemente por la hipoinsulinización relativa y el estado nutricional 49. En nuestra serie todos los niños, independientemente del tratamiento recibido, han presentado un crecimiento adecuado.
Concluyendo, nuestra serie muestra una tasa de complicaciones muy similar a la de otras casuísticas y revela, con objeto de prevenirlas, la importancia de un diagnóstico precoz y un tratamiento multidisciplinar por parte de equipos experimentados. En este sentido, creemos que la adquisición de mayor experiencia en el empleo de la tomografía de emisión de positrones para definir el tipo de lesión y optimizar el tratamiento quirúrgico mejoraría sustancialmente el tratamiento de estos pacientes. No obstante, todavía son necesarios estudios prospectivos multicéntricos a largo plazo que definan un modo de actuar consensuado, así como avances en genética molecular y terapia médica que mejoren definitivamente el pronóstico de estos pacientes.
Correspondencia: Dra. I. González Casado.
Hospital Universitario Infantil La Paz.
Servicio de Endocrinología Infantil.
P.º de la Castellana, 261. 28046 Madrid. España.
Correo electrónico: igonzalezc@hulp.insalud.es
Recibido en octubre de 2005.
Aceptado para su publicación en marzo de 2006.
