




Los hemangiomas infantiles (HI) son el tumor benigno pediátrico más frecuente; sin embargo, pueden presentar complicaciones como la ulceración, deterioro funcional, hipotiroidismo, o secuelas cosméticas, en cuyo caso el propranolol es el tratamiento de primera línea. Además, los HI pueden asociarse, en función de la localización, a otras entidades como el síndrome PHACE, LUMBAR o, en caso de haber 5 o más, a los hemangiomas hepáticos. Para descartarlos se recomienda el cribado con ecografía1.
Presentamos el caso de un lactante de 3 meses de edad con lesiones cutáneas asintomáticas, de aspecto vascular, presentes desde el nacimiento. Como antecedentes personales destacaban una gestación mediante técnicas de reproducción asistida, cesárea por sospecha de corioamnionitis subclínica, prematuridad (28+2), peso y talla elevados para la edad gestacional (percentil 92 y 98), hernia umbilical, leve macroglosia y enfermedad de membrana hialina tratada con una dosis de surfactante pulmonar. No presentaba antecedentes familiares de interés. A la exploración se objetivaron 7 lesiones papulosas eritemato-violáceas, de aspecto congestivo, de entre 2 y 6mm, localizadas en la palma derecha, pene, espalda, clavícula y hombro derecho, compatibles con HI múltiples (fig. 1).
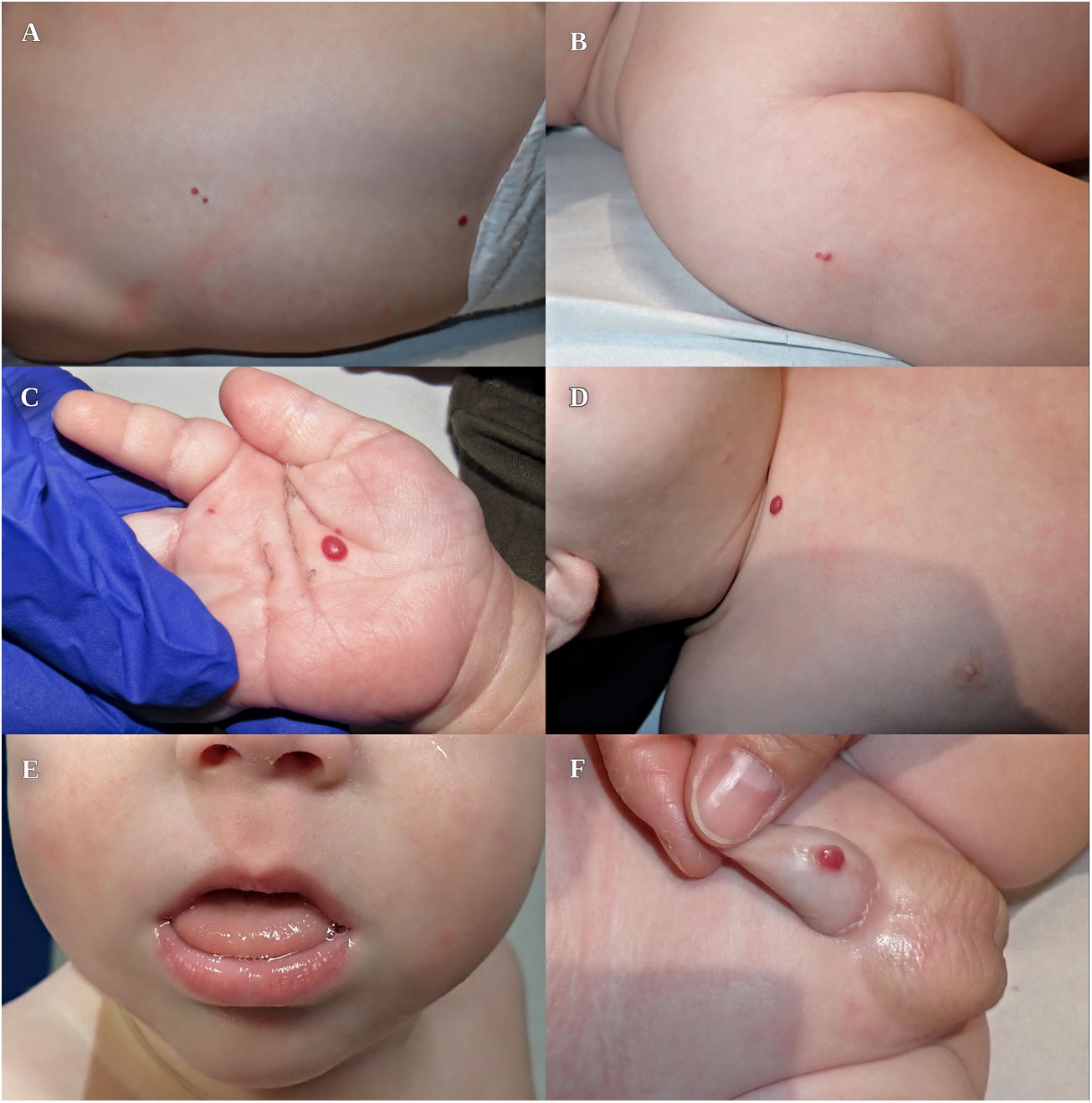
Debido a la presencia de más de 5 HI, se realizó una ecografía abdominal en la que se objetivaron entre 10 y 15 lesiones ocupantes de espacio, hipoecogénicas, ovaladas, algunas con señal doppler en su interior, compatibles con hemangiomas hepáticos (fig. 2 A y B). La analítica con perfil tiroideo, hepático y alfafetoproteína no mostró alteraciones relevantes. Se inició tratamiento precoz con propranolol 1mg/kg cada 12h por vía oral durante un año con desaparición de los hemangiomas hepáticos a los 4 meses (fig. 2 C y D) y disminución progresiva de los hemangiomas cutáneos. Dada la asociación de macroglosia, hernia umbilical y peso elevado al nacimiento, el paciente cumplía un criterio mayor y 2 menores (4 puntos totales) de síndrome de Beckwith-Wiedemann (SBW). Cuando la puntuación es mayor o igual a 4, se puede establecer un diagnóstico clínico de SBW sin necesidad de esperar el resultado genético, aunque siempre es recomendable realizarlo2. El estudio molecular de la región 11p15.5 objetivó una pérdida de metilación del dominio regulado por impronta genómica ICR2 (mediante análisis MS-MLPA), compatible con SBW.
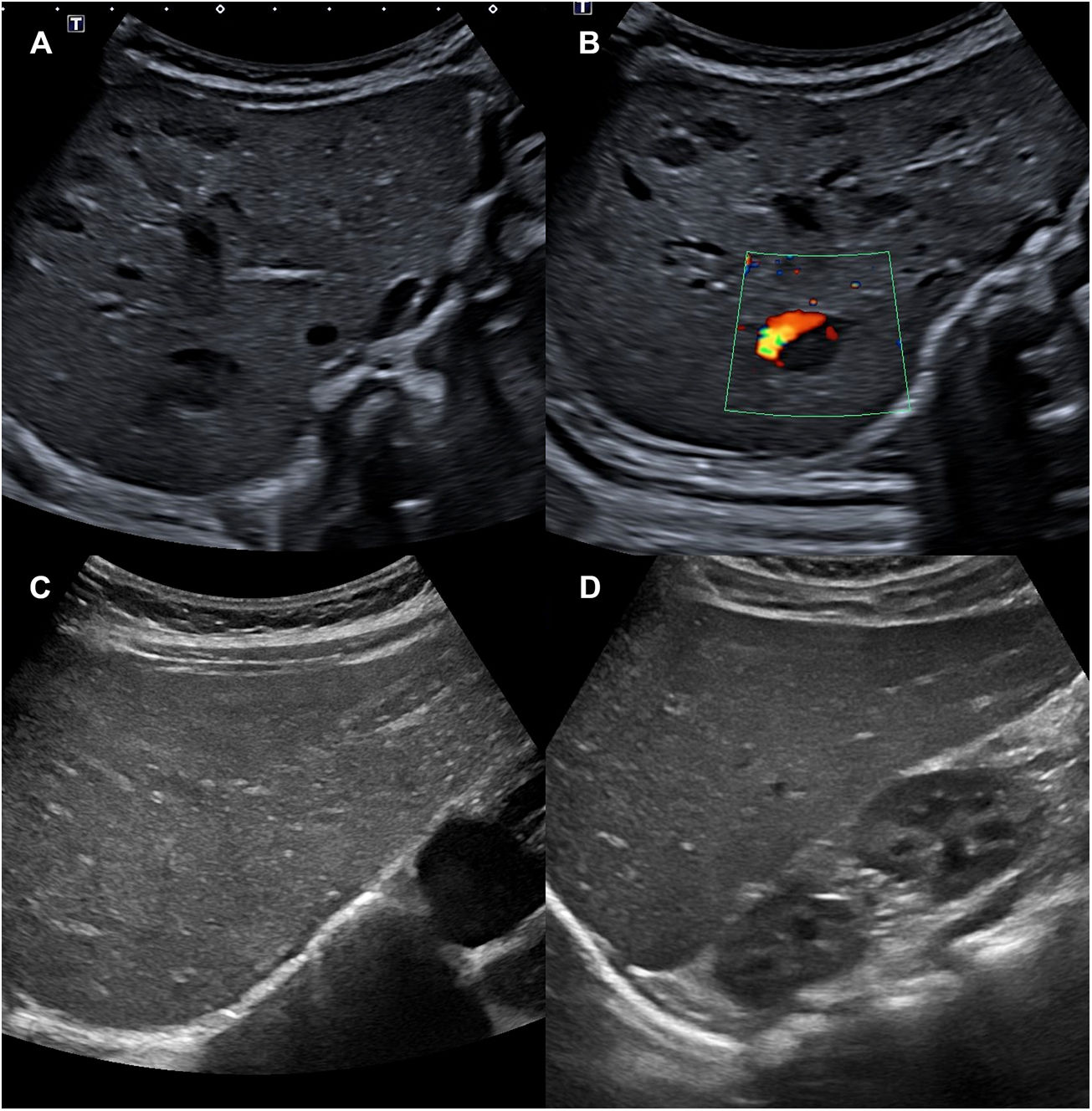
A y B) Ecografía abdominal en la que se visualizan entre 10 y 15 lesiones ocupantes de espacio hipoecogénicas compatibles con hemangiomas hepáticos. En la imagen B se aprecia señal doppler. C y D) Ecografía abdominal a los 4 meses de tratamiento, en la que han desaparecido los hemangiomas hepáticos. En la imagen no hay señal doppler.
El SBW es el síndrome de sobrecrecimiento más común, con una prevalencia de 1:10.340 en nacimientos concebidos de forma natural, y de 1:1.126 en caso de utilizar técnicas de reproducción asistida3. Se caracteriza por macroglosia, macrosomía, defectos de la pared abdominal, hipoglucemia neonatal y un mayor riesgo de tumores embrionarios2–6. Sin embargo, los tumores vasculares son muy poco frecuentes.
El SBW es causado mayoritariamente por errores en la impronta genómica (alteraciones epigenéticas) en la región 11p15.5 en dos dominios críticos, el centro de imprinting 1 y 2 (ICR1 e ICR2). El defecto causal más común (50% de los casos) es la pérdida de metilación en el dominio ICR2 en el alelo materno, lo que conlleva la inhibición de la expresión de CDKN1C, un gen implicado en la inhibición del ciclo celular. En menor frecuencia, puede ser secundario a una disomía uniparental paterna en la región 11p15 (20%); o secundaria a una ganancia de metilación en el alelo materno en el ICR1 (5%), que conlleva la sobreexpresión de IGF2 (factor de crecimiento tipo insulina 2) y represión de H19 (ARN no codificante supresor de tumores). Estas alteraciones dan lugar a los fenotipos de sobrecrecimiento asociados a este síndrome2–4.
Los HI se producen en aproximadamente el 4-5% de los recién nacidos, siendo el tumor benigno más común de la infancia1. Su patogénesis es poco conocida, aunque los últimos estudios demuestran un incremento de IGF2 en su fase proliferativa, pudiendo ser clave en la regulación de la proliferación de los hemangiomas7. La asociación de los HI con el SBW podría estar relacionada con el gen IGF27 y el CDKN1C, ya que ambos son fuertes reguladores del crecimiento fetal. En nuestro caso el defecto molecular identificado fue una pérdida de metilación del ICR2, lo que conlleva la inhibición de la expresión de CDKN1C, favoreciendo como resultado la proliferación celular.
Existen pocos casos descritos en la literatura que relacionen el SBW con HI. Recientemente, Macchiaiolo et al. informaron de la asociación de un paciente con SBW (disomía uniparental paterna) con hemangiomas cutáneos y hepáticos2,5. Previamente Francisco et al. habían publicado un caso de SBW con múltiples HI en pulmón, hígado y axila6. También ha habido informes previos de HI aislados en bazo, hígado, e incluso placenta en pacientes con SBW2. Hasta donde sabemos, esta es la segunda asociación reportada de SBW y HI múltiples cutáneos y hepáticos.
En conclusión, los tumores vasculares, en concreto los HI múltiples, podrían estar asociados con el SBW y pertenecer a su espectro, por lo que deben considerarse en el diagnóstico diferencial para un diagnóstico y tratamiento precoz.
