


Los forámenes parietales son pequeñas formaciones en torno al milímetro en la zona parasagital de los huesos parietales. Son una variante de la normalidad, presentes de manera uni o bilateral, hasta en un 60% de la población. Hablamos de agujeros parietales gigantes o foramina parietalia permagna (FPP) cuando miden más de 5mm de diámetro, con una incidencia mucho menor (1/15.000-25.000 personas)1. En realidad, la denominación es incorrecta, ya que los forámenes parietales pequeños pueden coexistir con los gigantes y no siempre es el agujero parietal en sí el que está alargado.
La FPP es un defecto de osificación primario a nivel de la eminencia parietal. En niños pequeños puede presentarse como una fontanela posterior permanentemente agrandada y posteriormente, durante la infancia, la línea media se osifica formándose las 2foraminas1.
La mayoría cursa de manera asintomática, aunque puede asociarse a cefalea, náuseas, vómitos o discapacidad intelectual y, en ocasiones, a otras malformaciones: craneosinostosis, displasias corticales, microcefalia, defectos oculares u óticos, disostosis craneofaciales, sindactilia, exóstosis múltiples o alteraciones de los vasos intracraneales1,2.
El 85% se debe a una mutación con herencia autosómica dominante y penetrancia alta pero incompleta (90%)1. La mayoría de las mutaciones descritas hasta el momento se encuentran en los genes MSX2 (5q35.2) y ALX4 (11p11.2). Ambas tienen una prevalencia similar y suelen ser clínicamente indistinguibles al tratarse de genes homeobox que codifican el homeodominio de ADN o ARN que luego dará lugar a la transcripción de factores implicados en el desarrollo esquelético3.
El diagnóstico se basa en los antecedentes familiares y el examen físico, pudiéndose complementar con pruebas de imagen: en radiografía simple se observan radiolucencias simétricas en la zona parasagital de los huesos parietales, en tomografía computarizada (TC) con reconstrucciones 3D pueden delimitar el defecto óseo y la resonancia magnética nuclear (RM) puede demostrar otras alteraciones intracraneales asociadas1. La confirmación diagnóstica es la detección de la mutación patógena con técnicas de genética molecular.
El tratamiento suele ser conservador, pudiéndose emplear medidas preventivas como ortesis craneales con el fin de evitar lesiones cerebrales. La cirugía quedaría reservada para forámenes de gran diámetro con riesgo de lesión: niños pequeños activos o con trastornos convulsivos, o aquellos casos que asocian otras malformaciones que la precisen1,4.
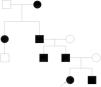
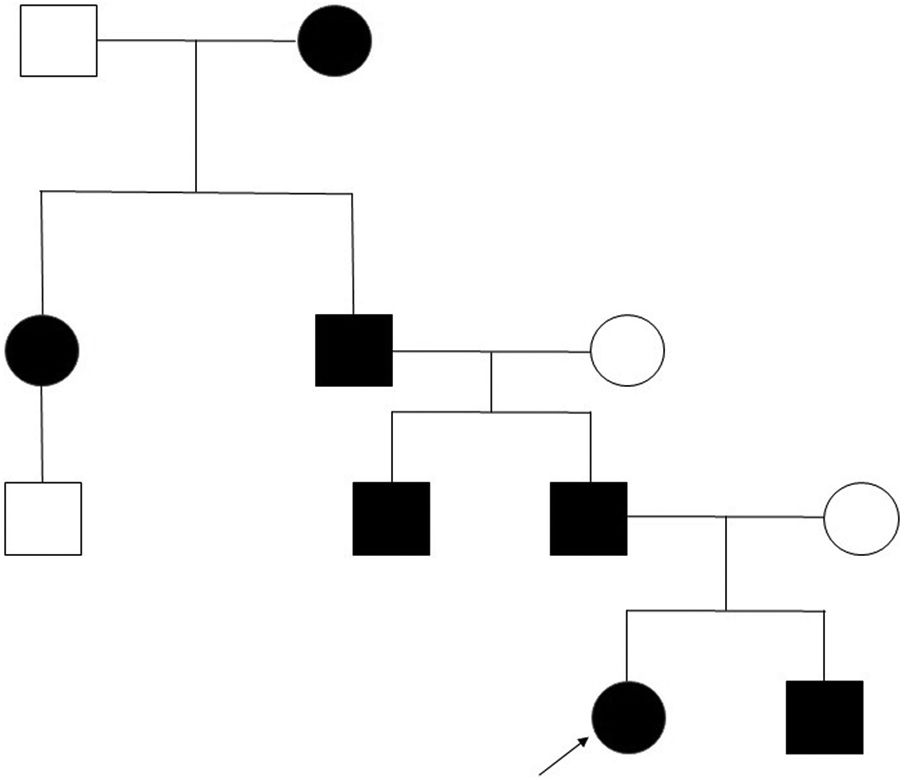
Caso clínico. Presentamos el pedigrí de una familia española con 7 individuos afectados de 4 generaciones consecutivas (fig. 1), diagnosticados de FPP portadores en heterocigosis de una mutación no descrita hasta la fecha del gen ALX4 (c.404_405dupG).
El caso índice es una recién nacida diagnosticada de un defecto de osificación craneal en ecografías prenatales por lo que se decide realizar cesárea electiva. Al nacimiento, presenta fontanela posterior extensa (5×5cm) sin otras malformaciones craneofaciales, fenotipo y exploración neurológica normal. En ecografía transfontanelar se descartan malformaciones asociadas, pero se aprecia una alteración de la ecogenicidad parietooccipital bilateral compatible con infartos de posible origen traumático, confirmados por RM craneal (fig. 2A). Tras 3 años de seguimiento, permanece asintomática con neurodesarrollo normal.
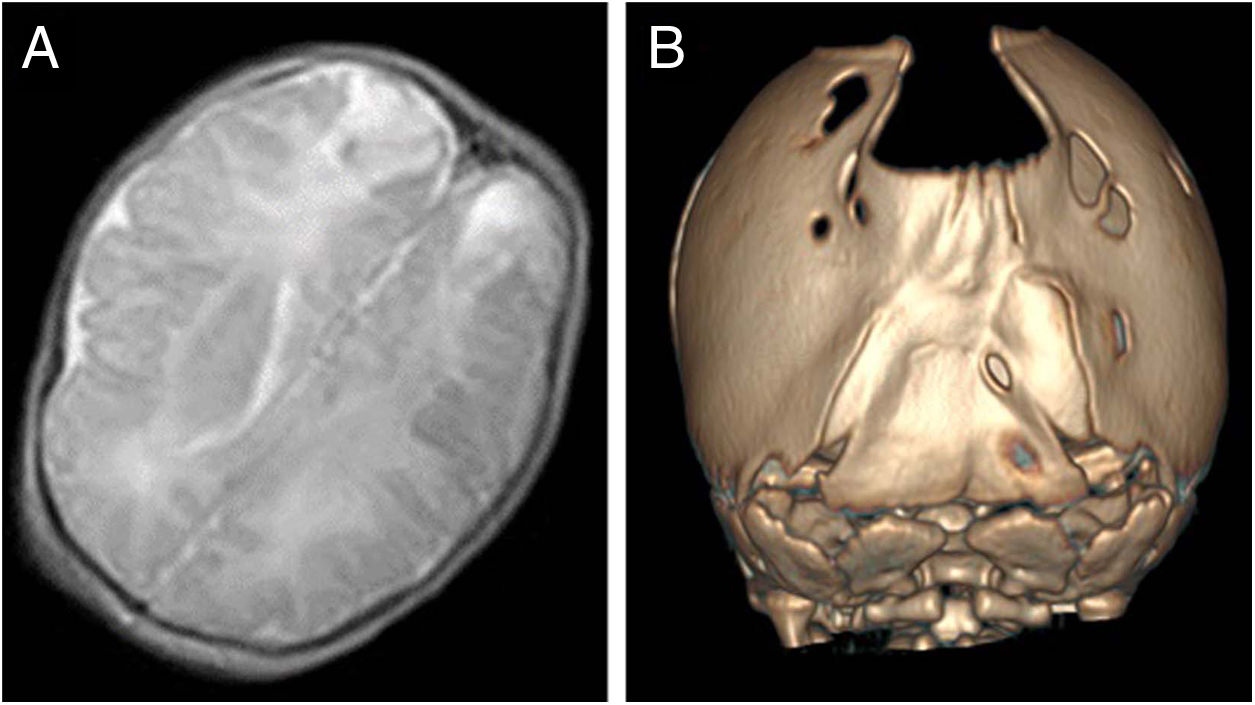
Su hermano menor fue diagnosticado intraútero con RM prenatal, donde se objetivó ausencia de componente óseo en línea media parieto-occipital. En la primera consulta de neuropediatría, se objetiva abombamiento frontal con aplanamiento occipital, se palpan crestas óseas en ambas suturas lamboideas y el defecto óseo parieto-occipital. Se realiza TC de calota craneal, diagnosticándose de craneosinostosis de ambas suturas lamboideas asociada a FPP (fig. 2B). Exploración neurológica es normal. Actitud quirúrgica para corrección de craneosinostosis.
El padre presentaba como único hallazgo en la exploración los agujeros óseos parietales característicos de esta entidad, sin asociar otros síntomas, al igual que los otros 4 miembros afectados de la familia.
La FPP es una entidad rara y de carácter benigno. El conocimiento de sus características clínicas y genéticas puede evitar la realización de exámenes complementarios y tratamientos innecesarios, disminuyendo la ansiedad de los padres, sobre todo si el diagnóstico es prenatal. Cuando se observen aperturas parietales bilaterales en ecografías prenatales, debería investigarse el pedigrí familiar para identificar familiares afectados que hayan pasado desapercibidos, debido a la variabilidad intrafamiliar o al posible cierre espontáneo del defecto óseo. La evaluación clínico-radiológica y la demostración de mutaciones en ALX4 o MSX2 en los familiares o el feto permitirá establecer un diagnóstico definitivo5,6. El defecto craneal de forma aislada podría no justificar la realización de un parto por cesárea, ya que esta, como en el caso índice presentado, no elimina definitivamente el riesgo de lesiones traumáticas secundarias al propio parto.
Presentado como póster en el 66.° Congreso AEP, Zaragoza, 7, 8 y 9 de junio del 2018.
