







Los errores congénitos del metabolismo son un grupo muy heterogéneo de enfermedades raras. La mayoría se pueden tratar con dieta y sustitución enzimática.
Existen pocos pacientes y pocos estudios publicados en estas enfermedades. Por ello, se ha llevado a cabo un estudio con el objetivo de evaluar la efectividad de los medicamentos huérfanos utilizados en errores congénitos del metabolismo de un hospital general de 630 camas.
Material y métodosEstudio descriptivo restrospectivo de 24 meses de duración en un hospital general de 630 camas. Se incluyeron los pacientes diagnosticados durante la infancia de errores congénitos del metabolismo y que acudieron a Hospital de Día o a la consulta de Farmacia.
ResultadosSe estudiaron 15 pacientes, con una edad media de 27,8 años, que recibieron tratamiento con nueve fármacos: sapropterina, fenilbutirato de sodio, miglustat, velaglucerasa, sebelipasa, idursulfasa, hidroxitriptófano, succinato y riboflavina. Presentaban siete errores congénitos del metabolismo diferentes: fenilcetonuria, trastorno del ciclo de la urea, Gaucher, Niemann-Pick, Hunter, déficit de lipasa ácida liposomal y defectos mitocondriales. Los medicamentos huérfanos para estos trastornos supusieron el 1,3% del gasto de farmacia. Algunos medicamentos huérfanos lograron que los pacientes estuvieran asintomáticos y otros, sólo produjeron una discreta mejoría. La efectividad de las fórmulas magistrales se evaluó mediante criterios subjetivos. En general, los pacientes presentaron buena tolerancia a estos tratamientos.
ConclusionesLos medicamentos huérfanos utilizados para errores congénitos del metabolismo presentan buen perfil de seguridad, pero gran disparidad en cuanto a su efectividad. Suponen un alto impacto económico.
La incertidumbre en cuanto a la evidencia clínica de los medicamentos huérfanos junto con el elevado coste, hace que la relación coste-efectividad sea controvertida.
Inborn errors of metabolism are a highly heterogeneous group of orphan diseases. Diet therapy and enzyme and coenzyme replacement are the most frequently used treatment. There are few patients and published studies about inborn errors of metabolism. The main objective of this study was to describe the effectiveness of orphan drugs in inborn errors of metabolism in paediatric patients.
Material and MethodsRetrospective descriptive study of 24 months on patients diagnosed with inborn errors of metabolism during childhood and who attended the pharmacy clinic or Day-Care Unit of a 630-bed general hospital.
ResultsThe study included 15 patients with a median age of 17.8 years and were treated with nine different drugs: sapropterin, sodium phenylbutyrate, miglustat, velaglucerase, sebelipase, idursulfase, 5-hydroxytryptophan, succinate, and riboflavin. Nine different inborn errors of metabolism were observed: phenylketonuria, defects of the urea cycle, Gaucher, Nieman-Pick, Hunter's disease, along with acid lipase deficiency, and mitochondrial diseases. Orphan drugs used for the treatment of inborn errors of metabolism accounted for 1.3% of hospital drug costs. Some orphan drugs achieved asymptomatic patients, but others just produced a modest symptomatic improvement. Most patients showed good tolerance to the treatment.
ConclusionsOrphan drugs used in inborn errors of metabolism had an easy to manage toxicity profile, with many disparities in effectiveness. These drugs have a high economic impact. The cost-effectiveness ratio for orphan drugs is a controversial issue due to their high cost and the inconclusive clinical evidence.
Las enfermedades raras (ER) son aquellas cuya prevalencia es inferior a cinco casos por cada 10.000 personas en la Comunidad Europea. La mayoría de los casos aparecen en la edad pediátrica, dada la alta frecuencia de enfermedades de origen genético y de anomalías congénitas. No obstante, la prevalencia es mayor en los adultos que en los niños, debido a la excesiva mortalidad de algunas enfermedades infantiles graves y a la influencia de ciertas enfermedades que aparecen a edades más tardías1.
Los errores congénitos del metabolismo (ECM) son un grupo amplio y heterogéneo de ER, con más de 500 tipos. Generalmente aparecen en las primeras etapas de la vida y la prevalencia global es de 1/600-1/1.000.
La mayoría son de origen genético, autosómicas-recesivas y suelen deberse a una alteración en la estructura o función de una proteína2.
El tratamiento habitual consiste en medidas dietéticas, medicamentos huérfano (MH) basados en la sustitución enzimática o ambos. La Comisión Europea define MH como «producto destinado a una indicación cuya prevalencia no exceda los cinco casos por cada 10.000 habitantes y usado para una enfermedad que ponga en riesgo la vida, o grave y crónica o sin ningún método satisfactorio de diagnóstico, prevención o tratamiento autorizado en la UE»3,4.
La baja prevalencia individual, la dispersión geográfica y los escasos estudios, en ocasiones con limitaciones metodológicas, dificultan el conocimiento sobre ER.
Por ello, parece importante estudiar las ER en la práctica clínica. El objetivo principal del estudio es evaluar la efectividad de los MH utilizados en ECM diagnosticados en edades pediátricas. Los objetivos secundarios tratan de analizar el perfil de seguridad y el impacto económico de los mismos.
Material y métodosEstudio descriptivo observacional retrospectivo realizado en un hospital general de 630 camas durante 2018 y 2019. Se incluyeron todos los pacientes con ECM, diagnosticados durante la infancia, que recogieron MH en consultas externas de farmacia o recibieron estos medicamentos en Hospital de Día.
Se recogieron datos demográficos (edad, sexo y ECM) y datos clínicos relacionados con la evolución de la enfermedad.
La variable principal del estudio fue la efectividad de los MH. Su evaluación se basó en los criterios de los ensayos clínicos de cada fármaco:
- •
Sapropterina: reducción de fenilalanina plasmática (< 360 nmol/mL) y mejora en la tolerancia a su ingesta5.
- •
Fenilbutirato de sodio: reducción del amoniaco plasmático (19-82 mcg/dL), así como disminución de crisis hiperamonémicas6.
- •
Velaglucerasa: normalización de los recuentos celulares (plaquetas y hemoglobina), volúmenes viscerales y biomarcadores, así como reducción de la infiltración ósea7.
- •
Miglustat en Gaucher: reducción de hepatoesplenomegalia8.
- •
Miglustat en Niemann-Pick tipo C: enlentecimiento de la enfermedad y estabilización o mejora de la deglución y deambulación8.
- •
Idursulfasa: reducción de glicosaminoglicanos (GAG) en orina y de visceromegalias y mejora de la resistencia (medida con la prueba de caminata de seis minutos: C6m) y la función pulmonar (medida con la capacidad vital forzada: CVF)9.
- •
Sebelipasa alfa: mejora de los parámetros lipídicos y hepáticos10.
- •
Fórmulas magistrales para defectos de la cadena mitocondrial: dado que no son fármacos autorizados y debido a la falta de ensayos clínicos, se evalúa la efectividad con criterios clínicos.
Para los objetivos secundarios se recogieron la seguridad de los MH y el coste del tratamiento anual.
Los datos se recogieron mediante el programa de dispensación farmacéutica y la historia clínica electrónica.
Se respetaron los principios éticos para la investigación médica en seres humanos promulgados en la Declaración de Helsinki11. De acuerdo con lo exigido por Ley Orgánica 3/2018, del 5 de diciembre, de Protección de Datos Personales y garantía de los derechos Digitales, la base de datos se realizó garantizando la confidencialidad de los pacientes12. El estudio recibió la aprobación del Comité Ético de Investigación con Medicamentos de La Rioja.
RESULTADOSSe seleccionaron 15 pacientes, el 60% hombres, con una edad media de 17,8 años (3-49) y siete ECM diferentes. El MH más prescrito fue sapropterina (n = 5; 33,0%) para el tratamiento de la fenilcetonuria. Las características principales de los ECM se describen en la tabla 113-21.
Características principales de los ECM encontrados en el estudio
| Patología | Enzima deficitaria | Producto acumulado | Herencia | Prevalencia | Manifestaciones clínicas | Tratamiento |
|---|---|---|---|---|---|---|
| FenilalaninemiaFenilcetonuria | Fenilalanina hidroxilasa ± cofactor BH4 | Fenilalanina | Autosómica recesiva | 1/10.000 nacidos vivos | Retraso mental, microcefalia, hipsarritmia, trastornos motores y alteraciones cutáneas | 1. Medidas dietéticas con control fenilalanina2. Tratamiento enzimático sustitutivo (TES) con tetrahidrobiopterina (BH4) en los casos respondedores.En los casos en los que el déficit sea de BH4 son necesarios precursores de L-dopa y 5 hidroxitriptófano |
| Trastorno del ciclo de la urea (TCU) | Enzimas que participan en la síntesis de urea | Amoniaco | Autosómica recesiva Deficit de ornitina transcarba-moilasa: ligada al X | 1/25-50.000 nacidos vivos | Las formas neonatales son casi siempre mortales, mientras que las formas tardías se manifiestan y evolucionan de manera variable. Aparece con frecuencia dolor abdominal y hepatomegalia, retraso del crecimiento y psicomotor, encefalopatía con trastornos conductuales, alucinaciones. | En el déficit de argininosuccinato sintetasa y argininosuccinato liasa, es posible estimular la parte restante del ciclo mediante un aporte de arginina. En los otros casos se puede utilizar fenilbutirato de sodio. |
| Nieman-Pick tipo C | Proteína del transporte de lípidos | Colesterol y esfingolípido | Autosómica recesiva, NPC 1 y 2 mutado | 1-9/100.000 nacidos | Hepatoespleno-megalia además de una afectación grave del SNC con síntomas psiquiátricos | Miglustat se aprobó en 2009 para su uso como tratamiento en ENP tipo C. |
| Gaucher | Beta-glucosidasa ácida | Glucosil-ceramida | Autosómica recesiva | 1/50.000-500.000 | -EG tipo 1: citopenias y hepatomegalia. Factor de riesgo cardiovascular.-EG tipo 2: forma neurológica aguda El TES no impide la progresión, mortalidad antes de 1 año-EG tipo 3:síntomas neurológicos lentos y progresivos | 1. Tratamiento enzimático sustitutivo (TSE): imiglucerasa y velaglucerasa (esta sólo indicada en EG tipo 1).2. Tratamiento por reducción de sustrato (TRS): miglustat y eliglustat.3. Trasplante de células madre: permite la curación, elevado riesgo. |
| Hunter (MPS tipo II) | Iduronato-2-sulfatasa | Dermatán sulfato y heparán sulfato (GAG) | Ligada al cromosoma X | 1/72.000-77.000 recién nacidos varones | Hernias, hepatoesplenomegalia, estatura baja, trastornos del comportamiento y regresión psicomotora que provoca déficit intelectual, sordera, trastornos cardiacos y respiratorios. En la forma atenuada, no hay alteración cognitiva | 1.Trasplante de progenitores hematopoyéticos (TPH)2.TES: forma purificada de la enzima iduronato-2-sulfatasa: Idursulfasa3. En estudio: terapia intratecal de TES |
| Déficit lipasa ácida liposomal (DLAL) | Enzima lipasa ácida lisosomal | Esteres de colesterol y triglicéridos | Autosómica recesiva, mutaciones en el gen LIPA | 1/350.000 nacimientos | Hepatomegalia y elevación de transaminasas, cLDL y TG en suero, en general con concentraciones bajas de cHDl. La mayoría desarrollan hepatopatía crónica y pueden presentar enfermedad cardiovascular | 1. Tratamientos de soporte: hipolipemiantes o trasplante hepático.2. TSE: En 2015 se aprobó LAL recombinante humana: Sebelipasa alfa. |
| Defecto cadena respiratoria | Sistema de fosforilación oxidativa (OXPHOS) | Mitocondrial, autosómica recesiva o autosómica dominante. A veces esporádico | 1:50.000 | Generalmente se presentan como trastornos multisistémicos. Se manifiestan principalmente en músculo esquelético, el sistema nervioso o la médula ósea (alto consumo de O2). La clínica más habitual es neuromuscular. | 1.Mejorar la función de la cadena respiratoria (riboflavina, ubiquinona)2. Facilitar la eliminación (carnitina, tocoferol).3. Prevenir el estrés oxidativo (retinol, ácido lipoico, vitamina C…).a) Trasplante de órganosb) Terapia génica: investigaciónc) Tratamiento de soporte: dieta, aporte vitamínico |
Cinco pacientes con una edad media de 16 años (11-24) recibían tratamiento con sapropterina. La mediana de dosis fue de 700 mg/día (300-900). La media de la fenilalanina plasmática fue 369,6 ± 157 nmol/mL. Dos pacientes mantuvieron niveles inferiores a 360 nmol/mL y pudieron seguir una dieta sin restricciones. El resto requirieron suplementación con nutrición enteral exenta de fenilalanina y mantuvieron niveles entre 400-550 nmol/mL.
Los pacientes realizaban una vida sin limitaciones aunque dos presentaron temblor en las extremidades.
Trastorno del ciclo de la urea: fenilbutirato de gliceriloUna mujer de 34 años fue diagnosticada con 11 meses de un déficit de ornitina transcarbamilasa. En 2009 inició fenilbutirato de sodio, sustituido en 2019 por fenilbutirato de glicerilo 3,5 g/8 h por motivos organolépticos. De manera concomitante, recibía una dieta hipoproteica y suplementos alimentarios de citrulina, carnitina, ácido fólico y calcio.
Inicialmente, el amonio plasmático osciló entre 40-100 μg/dL, niveles similares a los basales, alcanzando el máximo en 2017. Como manifestaciones clínicas presentaba encefalopatía severa con actitud autoagresiva y autista y sin comunicación verbal. En el último año, la paciente presentó dos crisis epilépticas por semana, aunque mostró una ligera mejoría de atención y concentración. Como efectos adversos, destacaron flatulencias e insomnio.
Enfermedad de Gaucher (EG): velaglucerasa y miglustatDos varones, de 19 y 48 años fueron diagnosticados de EG en la infancia. Ambos llevaban vida normal sin limitaciones.
Uno de ellos fue diagnosticado de EG tipo 3, tras iniciar al año de vida con hepatoesplenomegalia, malnutrición y retraso pondoestatural, en la deambulación y comunicación. Durante 10 años recibió imiglucerasa como terapia enzimática sustitutiva (TSE), que por su desabastecimiento, fue sustituida por velaglucerasa 60 U/kg quincenal en uso compasivo. Además, mantenía una dieta restrictiva en proteínas. Mediante TSE, consiguió mantener los niveles de hemoglobina y plaquetas dentro de la normalidad. Como manifestaciones clínicas presentaba alteraciones esqueléticas. El paciente refería malestar general tras la administración de velaglucerasa, por ello se sospechó de la presencia de anticuerpos contra la enzima.
El otro paciente, diagnosticado de EG tipo 1, fue esplenectomizado en 1998 y desde 2000 hasta 2003 recibió imiglucerasa. En 2007 fue incluido en un ensayo clínico con miglustat 100 mg/8 h, que continúa recibiendo en la actualidad como terapia por reducción de sustrato. Se alcanzaron los objetivos terapéuticos al año de tratamiento, consiguiendo reducir el volumen hepático más del 13% y mejorar la hemoglobina y las plaquetas en + 0,7 g/dL y + 8 × 109/L, respectivamente. Como efecto secundario presentó temblor de manos.
Niemann-Pick tipo C: miglustatUna niña de tres años padece Niemann-Pick tipo C neonatal, diagnosticada mediante confirmación genética. Fenotípicamente presentaba un importante diámetro abdominal y genu valgo bilateral. Fue diagnosticada de hipertensión portal por cavernoma, por lo que recibía espironolactona y propranolol. Inició miglustat tras el diagnóstico a dosis creciente hasta 100 mg/día. La paciente comenzó la deambulación independiente a los 27 meses y el lenguaje a los tres años, con buena capacidad de relación social. En la última ecografía presentaba una esplenomegalia sin relevancia clínica. Como efectos secundarios presentó diarreas y flatulencias.
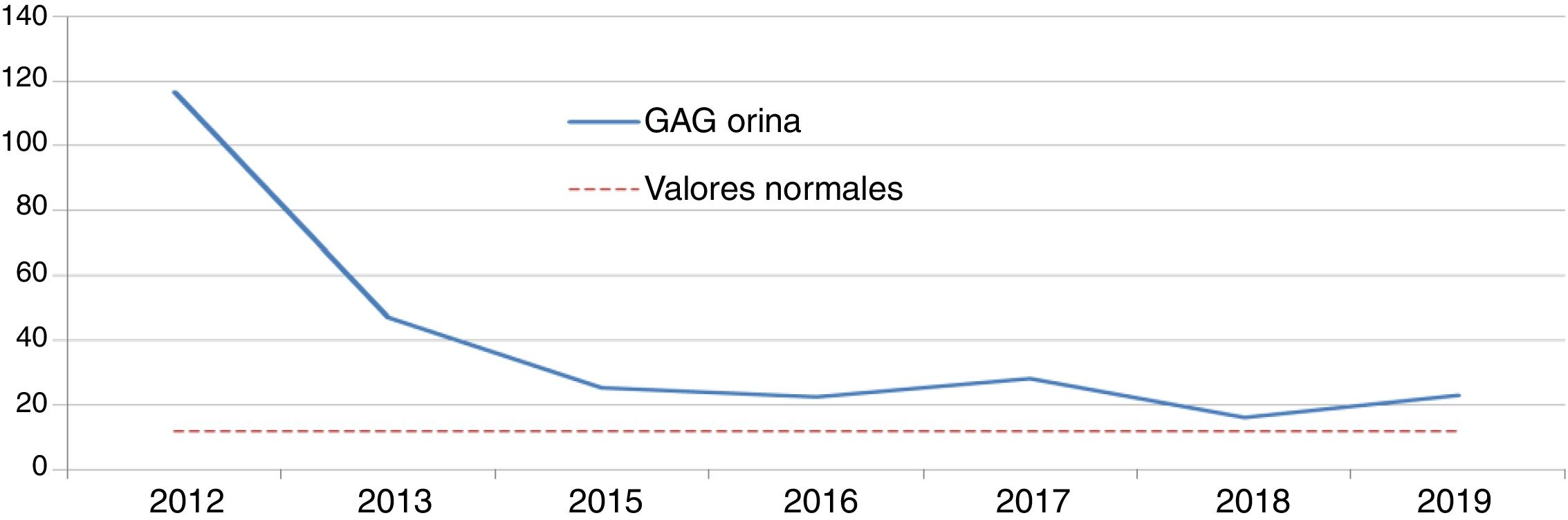
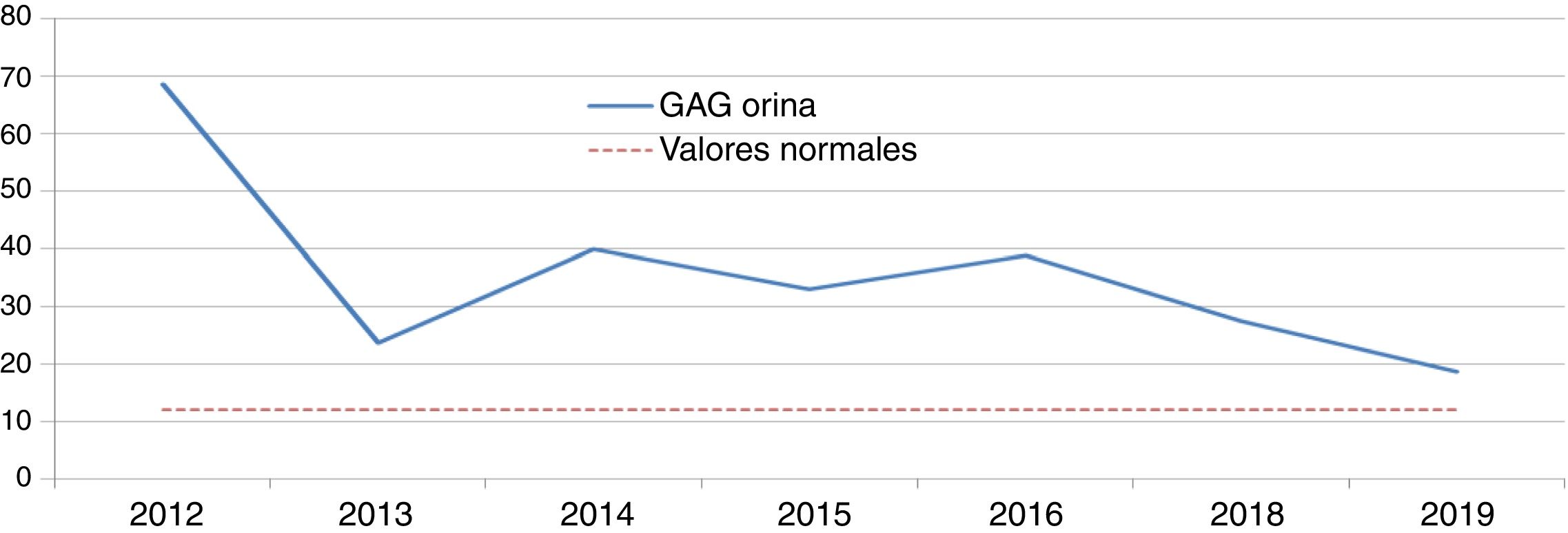
Enfermedad de Hunter (EH): idursulfasaDos niños de ocho y nueve años, fueron diagnosticados de EH por niveles de GAG en orina elevados y una actividad indetectable/mínima de iduronato-2-sulfatasa. Se confirmó el diagnóstico mediante análisis genético, presentando uno una mutación de novo y otro una familiar. Ambos pacientes recibían idursulfasa, uno a dosis semanal de 12 mg y otro a ciclos semanales de 12 mg-12 mg-18 mg. El descenso inicial de GAG en orina tras la instauración del tratamiento fue significativo; sin embargo, posteriormente los niveles se estabilizaron por encima de la normalidad (figs. 1 y 2). Uno de ellos presentó una evolución satisfactoria en el control de visceromegalias y función respiratoria, aunque estas variables fueron evaluadas mediante criterios clínicos subjetivos, sin llegar a realizar la C6m ni espirometría. Sin embargo, el otro, en la C6m logró caminar 300 metros sin disnea. Como manifestaciones clínicas, ambos tenían dificultades de aprendizaje con trastorno de atención, limitaciones de movimientos y pérdida en el control de esfínteres y del lenguaje. Clínicamente, uno presentaba hipoacusia bilateral, ametropía y neuropatía por atrapamiento; mientras que el otro presentaba comportamientos agresivos, estereotipias motoras, trastornos del sueño, aumento de apetito, insuficiencia mitral y aórtica sin repercusiones funcionales y cifosis. Acudía a un colegio de educación especial y realizaba actividades de ocio sin precisar apoyo ventilatorio, de alimentación o de deambulación. En ambos, las pruebas de imagen cerebral evidencian leucoencefalopatía y atrofia córtico-subcortical supratentorial. Además formaban parte de un ensayo clínico con terapia sustitutiva intratecal de forma concomitante. Como efectos adversos, uno presentó diarreas y frialdad de zonas acras y el otro desarrolló una taquicardia paroxística supraventicular.
Un paciente de 14 años fue diagnosticado genéticamente de DLAL en 2016, resultando doble heterocigoto. Desde los dos años ha presentado un déficit en peso (percentil 8) y talla (percentil 30). Estaba asintomático y realizaba vida normal, solo presentando bradicardia sinusal y hepatoesplenomegalia con esteatosis microvesicular. En 2018 inicia sebelipasa-alfa quincenal, actualmente a 35 mg, con lo que mejoró su perfil lipídico y hepático, sobre todo en los últimos meses (tabla 2).
Evolución de los perfiles lipídico y hepático en paciente con déficit de lipasa ácida liposomal
| Basal | Actual | % de variación | ||
|---|---|---|---|---|
| Perfil lipídico | Colesterol total (mg/dL) | 213 | 151 | -29,1 |
| Triglicéridos (mg/dL) | 153 | 110 | -28,0 | |
| c-HDL (mg/dL) | 37 | 39 | + 5,4 | |
| c-LDL (mg/dL) | 145 | 90 | -37,9 | |
| Perfil hepático | AST U/L | 123 | 48 | 61,0 |
| ALT U/L | 152 | 64 | 57,9 |
Dos mujeres, de 14 y 23 años, padecen un defecto en la cadena mitocondrial. Una de ellas tiene un déficit en el complejo III y IV de la cadena respiratoria, mientras que la otra es deficitaria en el complejo I.
Una presentaba dificultad motriz, hipotonía, crisis epilépticas y retraso en la comprensión y en el lenguaje. Ante la sospecha de un déficit de metabolitos de serotonina en el líquido cefalorraquídeo, se inicia tratamiento con hidroxitriptófano oral, 200 mg diarios, que se prepara como fórmula magistral en el servicio de farmacia. Actualmente, aunque presenta frecuentes caídas, ha mejorado en motricidad, comprensión y expresión del lenguaje.
La otra paciente comenzó, a los tres meses de edad, con anorexia, hipoacusia neurosensorial, comunicación interauricular y una dilatación biventricular cerebral. Como antecedentes familiares destaca el fallecimiento en los primeros seis meses de vida de cinco hermanos del padre por causas desconocidas. Le realizan una gastrostomía por dificultades para tolerancia oral. Actualmente está en tratamiento oral con riboflavina 60 mg cada 12 horas y succinato sódico un gramo cada 12 horas, que se preparan como fórmulas magistrales. Clínicamente, presenta un gran deterioro psicomotor e intelectual, sin comunicación verbal ni control de esfínteres. Es portadora de férulas para evitar autolesiones. Además tiene estreñimiento crónico, infecciones urinarias recurrentes y diversos problemas dentales.
Un niño de seis años comenzó al año de vida con trastornos en el desarrollo, presentando hipotonía muscular, dificultades para la succión, deglución y coordinación que le originaban problemas para la correcta nutrición. En 2015 diagnosticaron genéticamente, en el centro de referencia, una tetraparesia distónica sin filiar y retraso psicomotor. Sospechan de un defecto mitocondrial o un déficit de neurotransmisores. En el estudio de LCR destaca 5-hidroxitriptófano 45 nmol/L (valores normales: 0-26), 5-hidroxiindolacetico 11 nmol/L (valores normales: 170-440), ácido homovanílico 282 nmol/L (valores normales: 344-466), por lo que inician tratamiento con Dopa-Carbidopa y 5 hidroxitriptófano. Clínicamente presenta espasticidad en extremidades inferiores, trastorno del equilibrio, salivación profusa y dificultades ocasionales para la deglución. Ha mejorado en aspectos motores, consiguiendo deambulación con andador, así como en comunicación, lenguaje y manipulación. Está bien adaptado en el ambiente escolar. Ha presentado estreñimiento durante el tratamiento que ha impedido alcanzar las dosis plenas de hidroxitriptófano.
Análisis económicoLos resultados económicos de los MH para ECM se muestran en la tabla 3.
Coste de los medicamentos huérfanos utilizados para ECM
| Principio activo | Especialidad | Patología | Dosis | Precio mensual/ paciente | Precio anual/ paciente | Precio anual total(PVF €) |
|---|---|---|---|---|---|---|
| Sapropterina | Kuvan® 100 mg 120 comp | Hiperfenilalaninemia | 7 comp/ día* | 4.508 | 54.096 | 270.480 |
| Fenilbutirato de glicerilo | Ravicti® 1.1 g/mL 25 mL | Trastorno del ciclo de la urea | 3.5 g/8 h | 1.620 | 19.440 | 19.440 |
| Miglustat | Miglustat 100 mg 84 cap | Gaucher | 100 mg/8 h | 3.332 | 39.986 | 39.986 |
| Velaglucerasa | Vpriv® 400 UI | Gaucher | 4.260 UI/14 días | 25.740 | 308.880 | 308.880 |
| Miglustat | Miglustat 100 mg 84 cap | Nieman- Pick tipo C | 100 mg/24 h | 1.111 | 13.329 | 13.329 |
| Idursulfasa | Elaprase® 2 mg/ml 3 mL | Hunter | 12 mg/semanal* | 19.016 | 228.192 | 456.384 |
| Sebelipasa ácida | Kanuma® 2 mg/mL 10mL | Déficit lipasa ácida liposomal | 35 mg/14 días | 14.344 | 172.128 | 172.128 |
Los MH para ECM suponen un gasto anual de 1.290.674 euros, el cual representa aproximadamente un 3,0% del presupuesto farmacéutico del hospital (tomando como referencia el gasto de 2018). Los tratamientos de mayor coste por paciente son, por orden, velaglucerasa, idursulfasa y sebelipasa ácida, todos ellos, tratamientos intravenosos. Si hablamos del coste total, idursulfasa, seguido de velaglucerasa y sapropterina son los tratamientos con mayor impacto sobre el presupuesto. Los más asequibles son miglustat y fenilbutirato de glicerilo.
DiscusiónEl consumo de MH en España en el ámbito hospitalario en 2016 supuso el 8,4% del gasto hospitalario22, en concordancia con el de otros países europeos23. Los MH para ECM suponen un 1,3% del gasto farmacéutico del hospital, por lo que presumiblemente el coste de todos los MH sea superior a la media europea.
Considerando la prevalencia descrita de los ECM y la población de referencia del hospital, 300.000 personas aproximadamente, se pueden estimar los casos esperados en el estudio24. Los pacientes con Gaucher, Hunter y DLAL coinciden con los estimados. Por debajo se encuentran la fenilcetonuria, los trastornos del ciclo de la urea y Niemann-Pick. En estos dos últimos, la menor prevalencia puede deberse a un infradiagnóstico, al producirse la muerte en edades tempranas o porque no siempre necesitan un MH (dos de los seis defectos del ciclo de la urea se tratan exclusivamente con arginina)14.
En los ensayos clínicos de sapropterina se observó una media de 607 nmol/mL de fenilalanina tras seis semanas de tratamiento5. Nuestros pacientes tienen valores próximos al umbral terapéutico. Esto podría explicarse por la corta duración del ensayo, al contrario que en nuestro caso, donde los pacientes llevan años de tratamiento.
La eficacia de fenilbutirato de glicerilo en adultos se evaluó en dos estudios, donde los niveles de amonio plasmático se mantuvieron entre 10-51 μg/dL6. La menor efectividad de nuestro estudio podría deberse a que el paciente inició el tratamiento con signos avanzados de la enfermedad, mientras que en el ensayo los pacientes no debían tener ningún signo clínico de hiperamonemia. Entre otros efectos adversos, presentó insomnio, no descrito en la ficha técnica6.
El miglustat en Gaucher se aprobó tras un ensayo clínico de 28 pacientes. Se observó una reducción de la hepatomegalia además de un aumento medio de plaquetas de 22,2 × 109/L y de hemoglobina de 0,95 g/dL15. La efectividad de nuestro estudio fue ligeramente inferior, probablemente por la esplenectomía realizada, procedimiento no recomendado en la actualidad, que retrasó el inicio del tratamiento15.
La eficacia de velaglucerasa se evaluó mediante cuatro estudios que valoraban la normalización de los niveles de hemoglobina y de plaquetas. En un estudio de extensión se demostró una normalización del tamaño de hígado y bazo7. Nuestro paciente consiguió todos los objetivos terapéuticos, aproximándose los resultados a los obtenidos en los estudios.
La eficacia de miglustat en Niemann-Pick se evaluó en un ensayo clínico prospectivo y en una revisión restrospectiva, donde demostró disminuir los síntomas neurológicos relevantes8. Esta manera de evaluar la efectividad es subjetiva y por ello, sería necesario establecer medidas objetivas que permitan su valoración.
Se ha demostrado la seguridad y eficacia de idursulfasa en pacientes mayores de cinco años en dos estudios clínicos. La eficacia se midió con una variable compuesta, basada en la C6m y en la CVF9. No se realizaron espirometrías en nuestros pacientes por considerar el resultado no fiable en niños. Sólo se realiza C6m a uno de los pacientes, logrando un resultado superior al del ensayo clínico (43,3 metros). Convendría valorar si la realización de la prueba se hizo en las mismas condiciones que las del ensayo clínico. Como variables secundarias, se valoraron los GAG urinarios, que se normalizaron en el 50% de los pacientes y el control de visceromegalias25. En nuestros pacientes, el descenso inicial de GAG fue significativo, pero los niveles se estabilizaron al poco tiempo por encima de la normalidad. Ambos pacientes consiguen normalización de visceromegalias. En cuanto a la seguridad, uno de ellos presentó frialdad en las extremidades, no descrita en ficha técnica9.
Sebelipasa ácida demostró eficacia en un estudio multicéntrico. En nuestro caso, la reducción respecto al inicio de triglicéridos y c-LDL fue superior a la del ensayo, donde se obtuvieron unas reducciones de 25 y 28% respectivamente. El aumento de c-HDL fue inferior a la mejoría del 20% del ensayo. En el ensayo clínico se valoraron la normalización de los niveles de ALT, así como la reducción respecto al inicio que fue de un 53%10. Nuestro paciente, aunque con valores próximos al límite superior de normalidad no consigue la normalización, sin embargo presenta una reducción estimada del 58%. La efectividad muestra mejores resultados que la eficacia valorada en ensayos clínicos10. Esto podría deberse a que el perfil lipídico inicial de nuestro paciente era mejor que el de los pacientes del ensayo y que el periodo de duración del mismo fue de 20 semanas, frente al año y medio de seguimiento de nuestro estudio.
La efectividad y seguridad de las fórmulas magistrales usadas en los defectos de la cadena respiratoria se miden mediante criterios subjetivos. Parece necesaria la elaboración de guías o protocolos que evalúen los criterios clínicos a valorar para medir la efectividad de estas fórmulas en las diferentes patologías.
Es evidente que los ensayos clínicos presentan datos insuficientes sobre seguridad y eficacia, además de limitaciones en sus diseños: escaso número de pacientes, un único brazo, sin comparadores activos, pacientes con características demográficas muy diferentes, etcétera. Nuestro estudio también presenta una serie de limitaciones: es un estudio unicéntrico, con un tamaño muestral limitado, que no asegura una distribución representativa de la población. Además, puede existir sesgo de infrarregistro, ya que la historia clínica no está diseñada como fuente de datos. Sería necesario realizar estudios multicéntricos que incluyan un mayor número de pacientes y permitan estudiar los diferentes ECM por separado.
Se puede concluir que la efectividad de los MH para ECM es muy heterogénea. En algunos casos, como el fenilbutirato de glicerilo o idursulfasa suponen una discreta mejoría, mientras que otros suponen una estabilización clínica del paciente pudiendo incluso permanecer asintomático como en Gaucher.
En general, la tolerabilidad al tratamiento fue buena, a excepción de un paciente con idursulfasa que desarrolló una taquicardia paroxística supraventicular.
La incertidumbre en cuanto a la evidencia clínica de los MH junto con el elevado impacto económico, hace que la relación coste-efectividad sea controvertida.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



