

La realización de cualquier investigación con seres humanos o sus muestras biológicas requiere una valoración ética previa con el fin de evitar riesgos y problemas a todos los actores del proceso. La legislación al respecto es muy amplia, y hace hincapié en la preservación de los derechos de los pacientes en relación con los principios universales de autonomía, beneficencia y justicia. En este manuscrito se revisan las normativas sobre ensayos clínicos, estudios observacionales con medicamentos, proyectos de biobanco o cualquier otro tipo de estudio que se pueda realizar en el ámbito de la salud. Así mismo se analiza el papel de los comités de ética en investigación, la protección de datos y las bases de la integridad científica.
Any research conducted on human beings or human biological samples requires a prior ethical assessment to avoid risks and problems for all involved parties. The legislation in this regard is very broad and emphasizes the safeguarding of patient rights in relation to the universal principles of autonomy, beneficence and justice. The present article reviews the regulations applicable to clinical trials, observational studies with drugs, biobank projects or any other type of study that may be conducted in the health care field. It also addresses the role of research ethics committees, data protection and the foundations of scientific integrity.
La investigación biomédica debe realizarse conforme a los principios éticos universalmente aceptados que aseguran el avance del conocimiento, la mejora de la condición humana y el progreso de la sociedad, siempre respetando la dignidad del ser humano y la autonomía de su voluntad. Más recientemente se han incorporado aspectos como la protección de los datos de carácter personal, el bienestar animal y la preservación del medio ambiente1
No hace falta ir muy lejos en la historia de nuestra ciencia para comprender la necesidad de la ética en la investigación. Artículos como el de Beecher2 en 1966, o experimentos como el de Tuskegee realizado hasta 1972 en Alabama (EE. UU.)3, demuestran la necesidad de ser implacables en este sentido. La pediatría, como no podría ser de otra forma, tampoco ha sido ajena a los problemas éticos en investigación. El ejemplo del escándalo Willowbrook4 y el papel de Saul Krugman5 en esta controversia, se estudia en todos los cursos de bioética y es solo uno de los muchos ejemplos que se podrían citar.
Aunque nuestro mundo ha cambiado radicalmente desde la publicación de los principios éticos de la Declaración de la Asociación Médica Mundial en 1964, en Helsinki6, o el Informe Belmont en 19787, todavía hoy día es imprescindible que existan controles estrictos en cualquier investigación, por pequeña y banal que parezca, si no queremos caer en errores del pasado. Por este motivo, creemos de interés presentar en este artículo aspectos teóricos y prácticos relacionados con la ética en investigación y, de alguna manera, con la integridad científica en nuestra sociedad.
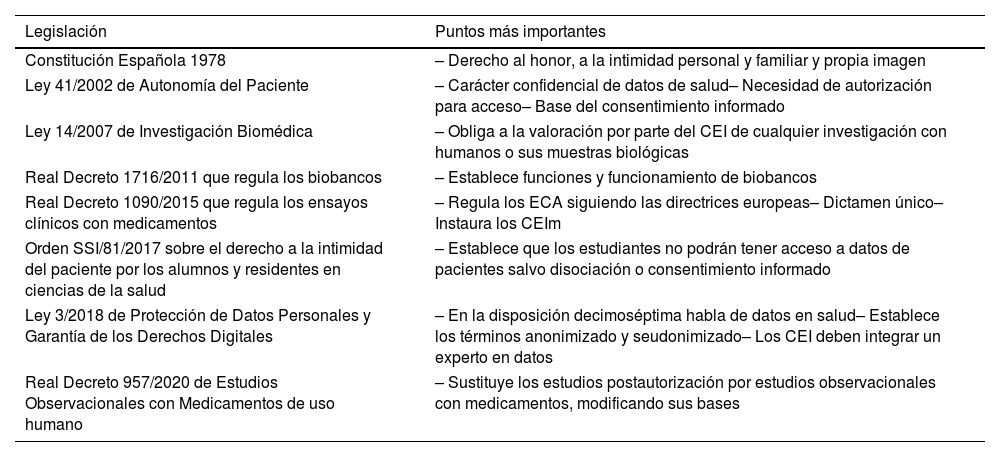
La legislación española sobre el temaLa legislación española sobre el tema es rica y variada, aunque algunos aspectos todavía están por desarrollarse (tabla 1). Aunque la Constitución Española ya habla del derecho a la intimidad personal, es la Ley 41/2002 de Autonomía del Paciente8 la que marca el principio de una serie de textos legales que van a ser de gran importancia en el devenir de la ética en investigación en España. Esta ley afirma que toda persona tiene el derecho a que se respete el carácter confidencial de sus datos de salud, y que nadie tiene derecho a acceder a ellos sin la previa autorización del sujeto, sentando las bases del concepto de consentimiento informado que será imprescindible en el futuro de la investigación biomédica.
Legislación española sobre ética en investigación
| Legislación | Puntos más importantes |
|---|---|
| Constitución Española 1978 | – Derecho al honor, a la intimidad personal y familiar y propia imagen |
| Ley 41/2002 de Autonomía del Paciente | – Carácter confidencial de datos de salud– Necesidad de autorización para acceso– Base del consentimiento informado |
| Ley 14/2007 de Investigación Biomédica | – Obliga a la valoración por parte del CEI de cualquier investigación con humanos o sus muestras biológicas |
| Real Decreto 1716/2011 que regula los biobancos | – Establece funciones y funcionamiento de biobancos |
| Real Decreto 1090/2015 que regula los ensayos clínicos con medicamentos | – Regula los ECA siguiendo las directrices europeas– Dictamen único– Instaura los CEIm |
| Orden SSI/81/2017 sobre el derecho a la intimidad del paciente por los alumnos y residentes en ciencias de la salud | – Establece que los estudiantes no podrán tener acceso a datos de pacientes salvo disociación o consentimiento informado |
| Ley 3/2018 de Protección de Datos Personales y Garantía de los Derechos Digitales | – En la disposición decimoséptima habla de datos en salud– Establece los términos anonimizado y seudonimizado– Los CEI deben integrar un experto en datos |
| Real Decreto 957/2020 de Estudios Observacionales con Medicamentos de uso humano | – Sustituye los estudios postautorización por estudios observacionales con medicamentos, modificando sus bases |
CEI: comité de ética en investigación; CEIm: comité de ética en investigación de medicamentos; ECA: ensayo clínico aleatorizado.
La segunda ley imprescindible en este tema es la Ley 14/2007 de Investigación Biomédica9 que, además de asegurar en su preámbulo que la investigación en salud es clave para mejorar la vida de los ciudadanos, obliga a que un comité de ética de la investigación (CEI) evalúe toda investigación biomédica que implique intervenciones en seres humanos o la utilización de sus muestras biológicas. De este modo, el control ético asignado a los comités en la legislación sobre ensayos clínicos se hace extensivo a toda la investigación biomédica. Esta ley señala que la investigación en humanos solo podrá llevarse a cabo en ausencia de alternativa experimental de eficacia comparable, que no deberá implicar riesgos y molestias desproporcionados en relación con los beneficios potenciales y que, cuando la investigación no tenga la posibilidad de producir resultados de beneficio directo para el sujeto participante, solo podrá ser iniciada en el caso de que represente un riesgo mínimo a juicio del CEI que deba evaluar la investigación.
Un Real Decreto (RD) transcendental en relación con la investigación biomédica es el RD 1716/2011 que regula los biobancos10. Este RD ordena todo lo referente al tratamiento, almacenamiento y circulación de muestras biológicas. Establece que las muestras biológicas de origen humano que vayan a ser almacenadas para investigación podrán ser de colecciones privadas, asociadas a un proyecto o de la colección del propio biobanco. Cada tipo de colección tendrá un manejo diferente, y para el manejo de las mismas los biobancos precisarán de un comité de ética externo para realizar la evaluación ética de las solicitudes de cesión, además de un comité científico externo que asesore los proyectos.
Pero sin duda ha sido el RD 1090/2015, por el que se regulan los ensayos clínicos con medicamentos (ECA), los comités de ética de la investigación con medicamentos (CEIm) y el Registro Español de Estudios Clínicos (REec)11, la norma que ha tenido un mayor impacto en el funcionamiento de los CEI. Se promulgó para dar respuesta a la exigencia del Reglamento (UE) 536/2014 y supuso un cambio de gran calado en el trabajo de los comités y la instauración del dictamen único. El RD explicita el papel de los CEIm en relación con la evaluación de los aspectos metodológicos, la idoneidad de las instalaciones, la adecuación de los investigadores principales, la hoja de información y el consentimiento informado, así como los aspectos relacionados con la protección de datos, la contratación de un seguro y la percepción de tasas por evaluación.
Siguiendo un orden cronológico, es importante señalar aquí la Orden SSI/81/201712 sobre el derecho a la intimidad del paciente por los alumnos y residentes en ciencias de la salud, que establece que los estudiantes no podrán tener acceso a los datos de los pacientes salvo previa disociación de estos a no ser que exista un consentimiento informado previo del paciente.
La Ley Orgánica 3/2018, de 5 de diciembre, de Protección de Datos Personales y Garantía de los Derechos Digitales (LOPD-GDD), dictada como desarrollo del Reglamento (UE) 2016/679 de Protección de Datos, ha regulado en su disposición adicional decimoséptima, el tratamiento en la investigación en salud13,14. Esta Ley establece el papel de los CEI en la evaluación de los estudios que utilizan datos relacionados con la salud, tanto identificados, seudonimizados o anonimizados, postulando nuevas exigencias en su composición, como la necesidad de contar con un delegado de protección de datos o, en su defecto, un experto del tema.
Por otro lado, el RD 957/2020 de Estudios Observacionales con Medicamentos (EOM) de uso humano15 sustituyó a la Orden SAS/3470/200916 que regulaba los Estudios Post-Autorización observacionales con medicamentos (EPA). Este RD no solo cambió el nombre de EPA por EOM, sino que modificó y simplificó los trámites de los estudios poscomercialización de los medicamentos, tan necesarios para asegurar la efectividad y seguridad de los medicamentos en condiciones de práctica clínica, eliminando la exigencia de clasificación previa de estos estudios por la AEMPS.
Para terminar este repaso, recientemente se ha aprobado el Reglamento del Comité Español de Ética de la Investigación17, organismo creado en 201118, adscrito al Consejo de Política Científica, Tecnológica y de Innovación, como órgano colegiado, independiente y consultivo, sobre materias relacionadas con la ética en la investigación y la integridad científica. Entre las funciones de este comité, se asigna la de establecer los principios generales para la elaboración de códigos de buenas prácticas de la investigación, que incluirán recomendaciones para prevenir, detectar, gestionar, evitar y resolver los conflictos de intereses.
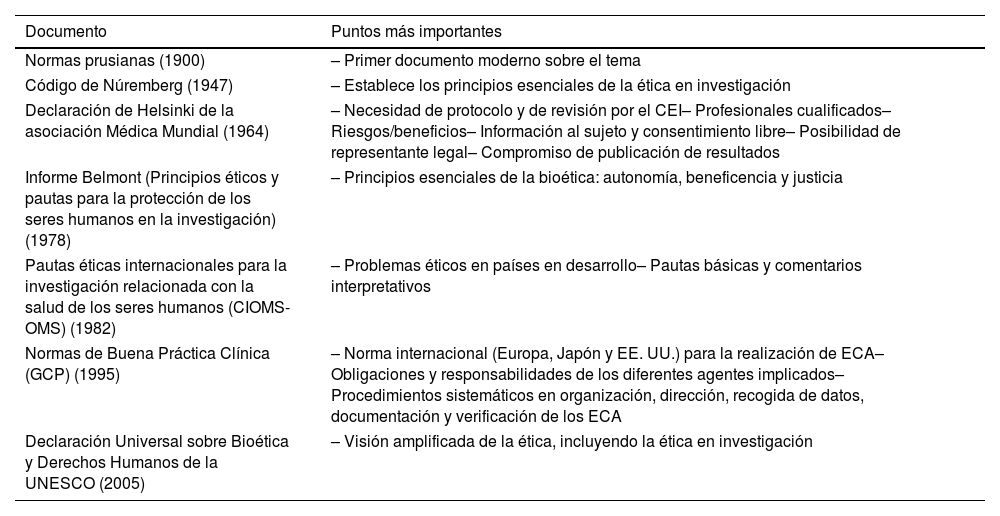
Documentos no legislativos/guías éticasLas normas prusianas, emitidas en 1900 a raíz del caso Neisser, se consideran el primer documento moderno sobre ética en investigación en occidente19,20. Posteriormente, el Código de Núremberg de 1947 fue el primero que señaló de forma clara y concisa los principios esenciales de la investigación: consentimiento voluntario, beneficio social, resultados previos que justifiquen el experimento, evitar sufrimiento físico o psíquico, valorar riesgo/beneficio, protección del paciente, investigadores cualificados, derecho a abandonar el experimento y posible suspensión del estudio si es necesario21. Sin embargo, el Código de Nuremberg presentaba limitaciones, como el hecho de que se refería a investigaciones con voluntarios sanos, priorizaba los intereses de la sociedad, establecía limitaciones para retirarse del estudio y no contemplaba la revisión ética externa. Se trataba de recomendaciones generales sin ningún rango legal.
La Asociación Médica Mundial, en 1964 emitió los principios éticos de la Declaración de Helsinki, que posteriormente fue modificada en sucesivas ediciones, siendo la última la de Fortaleza (Brasil) de 2013. La finalidad de la declaración fue proteger la dignidad de la persona y la intimidad de los participantes en la investigación. Estableció varios requisitos para que una experimentación fuese correcta, entre los que destacaban la necesidad de un protocolo experimental completo, revisado y aprobado por un comité de ética independiente, y que hubiera control y seguimiento del ensayo. Exigía también que los estudios fuesen dirigidos por profesionales cualificados y que existiera una ponderada relación entre riesgos y beneficios. Antes de que aceptase participar, el sujeto tendría que haber sido informado detalladamente de los procedimientos que conlleva la experimentación, los riesgos y cuantas circunstancias pudieran influir. El consentimiento se debía emitir con plena libertad y sin ningún tipo de coacción, presión o engaño. En el caso de personas sin plena capacidad para dar el consentimiento tendría que ser su representante legal el responsable de darlo en su lugar. Por último, exigía el compromiso de los investigadores en la publicación de los resultados, sean favorables o no.
En 1978 se publicó el Informe Belmont (Principios éticos y pautas para la protección de los seres humanos en la investigación) tras el escándalo del estudio de Tuskegee sobre sífilis en la población afroamericana, destapado por la prensa en EE. UU. En este documento se determinan los 3 principios esenciales de la bioética: respeto a las personas (autonomía), beneficencia y justicia7,22.
El principio de autonomía sostiene que todas las personan han de ser tratadas como sujetos autónomos, que aquellos que no lo sean han de ser protegidos, y que la autonomía varía a lo largo de la vida y puede necesitar ser reevaluada. La autonomía es la esencia de la necesidad de informar sobre el estudio y solicitar el consentimiento informado. La beneficencia, y su posterior desarrollo en el principio de no maleficencia, habla de evaluar siempre los riesgos y beneficios, maximizando estos últimos. Este principio es la razón para que se valore siempre la metodología y el diseño de los estudios en los CEI. Por último, el principio de justicia habla de la equidad en la selección de los participantes de los estudios, así como en la distribución de los riesgos y beneficios esperados23,24.
«Pautas éticas internacionales para la investigación relacionada con la salud de los seres humanos» fue un documento publicado por el Council For International Organizations Of Medical Sciences (CIOMS), en consenso con la Organización Mundial de la Salud (OMS) en 1982. Estas guías, cuya cuarta edición se publicó en 2016, abordaronn los temas éticos de investigación característicos de los países en desarrollo, con 25 pautas básicas y sus comentarios interpretativos25,26.
Otro documento imprescindible fue la Declaración Universal sobre Bioética y Derechos Humanos de la Organización de las Naciones Unidas para la Educación, la Ciencia y la Cultura (UNESCO) de 2005, que abogaba por respetar los principios de dignidad y derechos humanos, la autonomía, la vulnerabilidad y la integridad, la privacidad y confidencialidad, la igualdad, justicia y equidad, el respeto a la diversidad, la responsabilidad social, la solidaridad y cooperación, la protección de generaciones futuras, y la protección del medio ambiente27.
Para terminar, es imprescindible citar las Normas de Buena Práctica Clínica (Good Clinical Practice [GCP]), de la International Conference on Harmonitation (ICH). Se trata de una normativa internacional de calidad ética y científica aplicable al diseño, realización, registro y comunicación de los ensayos clínicos en los que participen seres humanos. Su cumplimiento asegura la protección de los derechos, seguridad y bienestar de los participantes en el ensayo y garantiza la fiabilidad de los resultados. Es importante destacar, dentro de las guías ICH para la realización de ensayos clínicos, la E11 destinada a poblaciones pediátricas28,29.
En la tabla 2 se puede leer muy resumidos los documentos no legislativos más importantes de este tema.
Documentos no legislativos más importantes sobre ética en investigación
| Documento | Puntos más importantes |
|---|---|
| Normas prusianas (1900) | – Primer documento moderno sobre el tema |
| Código de Núremberg (1947) | – Establece los principios esenciales de la ética en investigación |
| Declaración de Helsinki de la asociación Médica Mundial (1964) | – Necesidad de protocolo y de revisión por el CEI– Profesionales cualificados– Riesgos/beneficios– Información al sujeto y consentimiento libre– Posibilidad de representante legal– Compromiso de publicación de resultados |
| Informe Belmont (Principios éticos y pautas para la protección de los seres humanos en la investigación) (1978) | – Principios esenciales de la bioética: autonomía, beneficencia y justicia |
| Pautas éticas internacionales para la investigación relacionada con la salud de los seres humanos (CIOMS-OMS) (1982) | – Problemas éticos en países en desarrollo– Pautas básicas y comentarios interpretativos |
| Normas de Buena Práctica Clínica (GCP) (1995) | – Norma internacional (Europa, Japón y EE. UU.) para la realización de ECA– Obligaciones y responsabilidades de los diferentes agentes implicados– Procedimientos sistemáticos en organización, dirección, recogida de datos, documentación y verificación de los ECA |
| Declaración Universal sobre Bioética y Derechos Humanos de la UNESCO (2005) | – Visión amplificada de la ética, incluyendo la ética en investigación |
GCP: Good Clinical Practice; CEI: Comité de ética en investigación; CIOMS: Council For International Organizations Of Medical Sciences; ECA: ensayo clínico aleatorizado; OMS: Organización Mundial de la Salud; UNESCO: Organización de las Naciones Unidas para la Educación, la Ciencia y la Cultura.
El origen de los CEI en España se remonta a la segunda mitad del siglo pasado. Aunque en algunos centros hospitalarios ya existían grupos que, de alguna forma, se encargaban de valorar los aspectos éticos, la primera constancia legislativa en nuestro país de su existencia se encuentra en el RD de 1978 (RD 944/1978)38 y desde entonces han sido varios reales decretos y órdenes ministeriales que han ido delimitando sus funciones, estructura, legislación y denominaciones. Pero fue la Ley de Investigación Biomédica de 20079 la que estableció la necesidad de aprobación de cualquier estudio de investigación con humanos o sus muestras biológicas por los CEI.
El RD 1090/2015 define los CEI como órganos independientes de composición multidisciplinar cuya finalidad principal es la de velar por la protección de los derechos, seguridad y bienestar de los sujetos que participen en una investigación biomédica y ofrecer garantías mediante un dictamen sobre la documentación del proyecto, teniendo en cuenta los puntos de vista de personas legas, en particular los pacientes o las organizaciones de pacientes.
Este RD 1090/2015 definió el papel de los CEIm (CEI con poder para aprobar ECA de medicamentos), así como su estructura y funcionamiento, estableciendo que deben ser acreditados por la autoridad sanitaria competente, añadiendo por otro lado que ni el CEIm en su conjunto ni ninguno de sus miembros podrán percibir directa ni indirectamente remuneración alguna por parte del promotor del estudio. Por otra parte, en relación con la evaluación de ECAs con menores, el CEIm debe contar entre sus miembros con expertos en pediatría o haber recabado asesoramiento sobre las cuestiones clínicas, éticas y psicosociales en el ámbito de la pediatría.
La labor de estos comités (ya sean CEI o CEIm) radica en mantener el rigor científico y metodológico, verificar el cumplimiento de la normativa y los principios éticos, así como garantizar el bienestar y seguridad de los participantes en la investigación. Su función también recoge evaluar las modificaciones relevantes de los estudios autorizados y hacer un seguimiento de estos hasta la recepción del informe final. Aunque inicialmente la normativa solo hacía referencia a la evaluación de ensayos clínicos (ECA), actualmente sí explicita la función de evaluar cualquier estudio con seres humanos, sus datos de salud o su material biológico, tengan el diseño y/o complejidad que tengan.
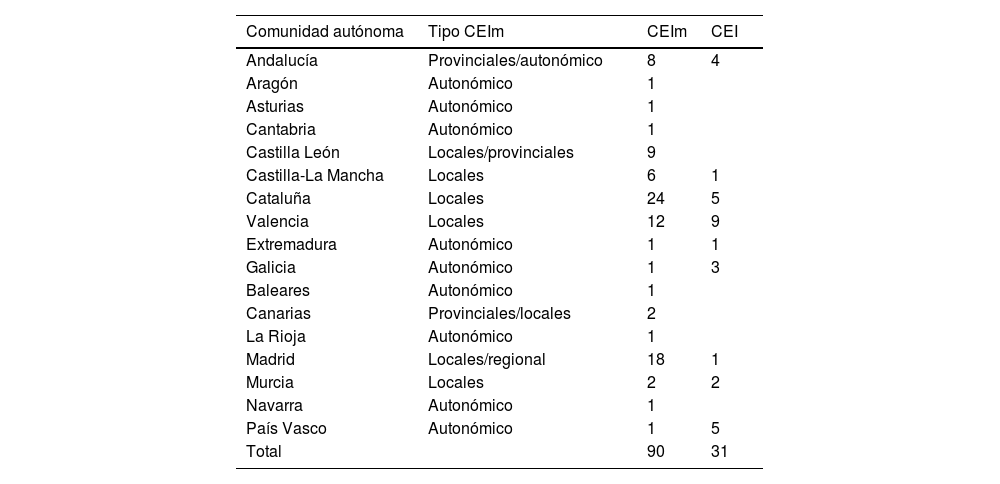
Según el registro de la AEMPS del 2021, existen 90 CEIm acreditados en nuestro país39 (tabla 3). El ámbito de actuación de los CEIm es muy variado, existiendo algunos autonómicos, otros de ámbito provincial o similar, y otros de ámbito hospitalario o institucional. Los CEIm son los únicos comités que están acreditados para evaluar estudios con medicamentos y/o productos sanitarios, mientras que los CEI pueden evaluar el resto de tipos de proyectos.
Comités de ética en investigación (CEIm y CEI) en Esapaña (registro de la Agencia Española del Medicamento y Productos Sanitarios)
| Comunidad autónoma | Tipo CEIm | CEIm | CEI |
|---|---|---|---|
| Andalucía | Provinciales/autonómico | 8 | 4 |
| Aragón | Autonómico | 1 | |
| Asturias | Autonómico | 1 | |
| Cantabria | Autonómico | 1 | |
| Castilla León | Locales/provinciales | 9 | |
| Castilla-La Mancha | Locales | 6 | 1 |
| Cataluña | Locales | 24 | 5 |
| Valencia | Locales | 12 | 9 |
| Extremadura | Autonómico | 1 | 1 |
| Galicia | Autonómico | 1 | 3 |
| Baleares | Autonómico | 1 | |
| Canarias | Provinciales/locales | 2 | |
| La Rioja | Autonómico | 1 | |
| Madrid | Locales/regional | 18 | 1 |
| Murcia | Locales | 2 | 2 |
| Navarra | Autonómico | 1 | |
| País Vasco | Autonómico | 1 | 5 |
| Total | 90 | 31 |
CEI: comité de ética en investigación; CEIm: comité de ética en investigación de medicamentos.
En 2011 se creó en España la Asociación Nacional de Comités de Ética en Investigación (ANCEI)40 que mantiene una actividad relevante en lo referente a colaboraciones institucionales, representación y docencia. ANCEI colabora con los CEI/CEIm en su labor, fomentando el trabajo en equipo y elaborando materiales de ayuda que publica periódicamente.
Manejo de datos personalesLa protección de la privacidad del sujeto a estudio (imagen, intimidad y honor) y la confidencialidad del manejo de sus datos por parte del investigador son dos aspectos esenciales en la realización de los proyectos de investigación en el ámbito sanitario.
La ley de Autonomía del Paciente8 limita el uso de los datos relacionados con la salud contenidos en la historia clínica a la asistencia; siendo necesario el consentimiento del paciente para la utilización de esta información para otros fines, incluida la investigación. El consentimiento informado, por lo tanto, es esencial en la ética de la investigación, como garantía del cumplimiento del principio de autonomía.
Sin embargo, como se comenta más adelante, el acceso a la historia clínica con fines de investigación ha sido modificado con la entrada en vigor de la LOPD-GDD, incluyendo los supuestos de investigación previstos en esta Ley entre las excepciones permitidas al acceso sin consentimiento a la historia clínica.
El Reglamento Europeo (UE) 2016/679 de protección de datos (RGPD) establece los principios que debe regir el tratamiento de datos relativos a la salud, permitiendo con carácter general su utilización con fines científicos. Pero, aunque se permita dicho uso, esta utilización debe ser proporcional a los objetivos planteados, respetar el derecho a la protección de datos y establecer medidas adecuadas para proteger los derechos e intereses de los sujetos.
La LODP-GDD13, recoge los principios del RGPD, haciendo referencia a la exigencia de licitud y transparencia en el tratamiento de los datos, la limitación en el uso por su finalidad, la minimización y exactitud de los datos, la limitación en el tiempo de conservación y la integridad y confidencialidad. Dedica el segundo apartado de la Disposición adicional decimoséptima al tratamiento de datos en la investigación en salud, exigiendo para estos tratamientos la aprobación previa por un CEI y estableciendo ciertos requisitos (tabla 4).
Requisitos de la Ley de Protección de Datos Personales y garantía de los derechos digitales (LODP-GDD 2018) en el tratamiento de datos de salud para investigación biosanitaria
| 1. Consentimiento | El interesado (o su representante legal) podrá otorgar el consentimiento para el uso de sus datos con fines de investigación en salud. |
| 2. Exención del consentimiento en situaciones excepcionales | Las autoridades sanitarias e instituciones públicas con competencias en vigilancia de la salud pública podrán llevar a cabo estudios científicos sin consentimiento de los afectados en situaciones de excepcional relevancia y gravedad para la salud pública. |
| 3. Reutilización de datos | Se considerará lícita y compatible la reutilización de datos personales con fines de investigación en salud cuando se utilicen para finalidades o áreas de investigación relacionadas con el estudio inicial. |
| 4. Seudonimización | – Se debe cumplir la separación técnica y funcional entre quienes la realicen y el equipo investigador.– Los datos seudonimizados conllevan compromiso de confidencialidad y de no reidentificación.– Debe evitarse el acceso a terceros.– Solo se permitirá reidentificación en caso de problema de salud o seguridad del sujeto, o amenaza grave a sus derechos, o para garantizar su asistencia sanitaria. |
Como novedad, la utilización de datos seudonimizados habilita el acceso a la historia clínica con motivos de investigación sin el consentimiento del sujeto. Esta exención de consentimiento se debe solicitar de forma justificada a los comités, que deberán valorar en cada caso si se cumplen los requisitos establecidos. Los comités podrían admitir también esta exención en estudios retrospectivos en los que el esfuerzo de obtener los consentimientos sea excesivo para la realización del estudio (fallecimientos, cambios de domicilio, …) y cuyas pérdidas puedan suponer un sesgo en los resultados, siempre que no se pueda proceder a la seudonimización o anonimización de los datos.
En relación con la realización de ensayos clínicos en pediatría es necesario tener en cuenta la necesidad de disponer de información contrastada que evite el uso de medicamentos fuera de indicación y el uso de medicamentos fuera de ficha técnica41-43. En este sentido debe destacarse la exigencia de la Ley de investigación biomédica que establece «se otorgará el consentimiento por representación cuando la persona esté incapacitada legalmente o sea menor de edad, siempre y cuando no existan otras alternativas para la investigación» y que «las personas incapacitadas y los menores participarán en la medida de lo posible y según su edad y capacidades en la toma de decisiones a lo largo del proceso de investigación».
Integridad científica y códigos de buenas prácticas científicasLa Declaración Nacional sobre Integridad Científica firmada por la Confederación de Sociedades Científicas de España (COSCE), la Comisión de Rectores de Universidades Españolas (CRUE) y el Centro Superior de Investigación Científica (SCIC) de 2015 señala que la integridad científica se relaciona directamente con el rigor científico, la excelencia y calidad de la investigación, y con un comportamiento ético y responsable en el quehacer científico30. Por lo tanto, nos referimos a integridad científica cuando hablamos de principios, valores y responsabilidades que son la base de la buena práctica científica. La Declaración de Singapur de 201031 establece 4 principios básicos: honestidad en todos los aspectos de la investigación, responsabilidad en la realización de ésta, cortesía profesional e imparcialidad en las relaciones entre científicos, y buena gestión de la investigación y sus recursos.
La integridad científica debe estar presente en todos los pasos del proceso científico, desde la pregunta a la comunicación de los resultados, pasando por el planteamiento metodológico, ejecución y evaluación de resultados, sin olvidarnos que trasciende al ámbito laboral, tanto en la interacción entre profesionales como en su formación. La integridad es una obligación moral compartida por todos los actores del proceso, incluidos investigadores, administración, instituciones, entidades financiadoras, editores y sociedades científicas.
Para mantener la integridad científica se han promulgado los llamados códigos de buenas prácticas científicas que, sin sustituir a la legislación, son directrices que afianzan los estándares éticos, favorecen la calidad de la investigación y previenen la mala práctica. Prácticamente todas las sociedades o instituciones relacionadas con la investigación tienen su propio código de buenas prácticas científicas, siendo el Código de All European Academies (ALLEA)32 el más representativo.
En relación con las buenas prácticas, podemos señalar la necesidad de mantener una política impecable de declaración de conflictos de intereses por parte de todos los implicados. Los conflictos de intereses son situaciones en las que el juicio de un profesional relacionado con un interés primario (integridad) puede estar indebidamente influenciado por un interés secundario. Estos conflictos deben ser declarados y no se consideran mala práctica si se identifican y se tienen en cuenta. Existen muchos tipos de conflictos de intereses, no solo económicos, y se debe apostar por la transparencia. Los investigadores, pero también los académicos, editores de revistas, evaluadores, personal de la administración y miembros de comités deben hacer declaración pública de sus intereses para que no haya duda de su independencia e integridad33,34.
La mala praxis científica no solo recae en la triada clásica de plagio, fabricación y falsificación, sino que también se puede dar en toda la escalera de la producción científica. En este sentido, por ejemplo, la integridad de los miembros de los equipos editoriales de las revistas científicas es esencial a la hora de la difusión y publicación de resultados35–37.
Si bien es cierto que las funciones asignadas a los CEI hacen referencia explícitamente a la evaluación, la emisión del dictamen de los proyectos de investigación y el seguimiento de las condiciones de realización de los mismos, no puede obviarse la función docente y su papel en garantizar la integridad científica de la investigación, tal y como refleja la Ley de investigación biomédica, En este sentido, debe destacarse la labor que los comités desempeñan en la difusión de los aspectos éticos y legales de la investigación biomédica dirigida a investigadores para garantizar el cumplimiento de los estándares éticos en investigación.
A modo de conclusiones prácticasTodo estudio con seres humanos o sus muestras biológicas debe ser aprobado por un CEI/CEIm. Esto atañe tanto a los estudios de gran calado (ensayos clínicos, estudios observacionales de medicamentos, estudios con muestras biológicas y/o estudios con dispositivos o productos sanitarios), como a todo tipo de proyecto en el que se manejen datos de pacientes, sean del tipo, importancia y propósito que sean, desde los trabajos de fin de estudios hasta las investigaciones observacionales retrospectivas que se realizan con ánimo de publicar o comunicar sus resultados.
La aprobación previa de estos estudios por un comité es imprescindible porque asegura el cumplimiento de los principios éticos y el respecto legal a los derechos de los participantes, así como su calidad metodológica y la seguridad de su realización, partiendo de la premisa de que si un estudio es metodológicamente incorrecto no será éticamente aceptable.
La solicitud de consentimiento informado a los pacientes debe ser la norma de todo estudio de investigación. La protección de la privacidad de los datos de los pacientes es esencial en todo el proceso y, aunque existen situaciones en las que se puede solicitar la exención, el consentimiento informado es la pieza clave en garantizar el cumplimiento ético del principio de autonomía. No obstante, la valoración de la solicitud de exención del consentimiento debe ser estudiada por un CEI/CEIm, que decidirá su dictamen tras comprobar las circunstancias del estudio.
Los CEI/CEIm tienen un papel clave en el fomento y la promoción de los aspectos éticos y legales de cara a garantizar la integridad científica de la investigación, al promover valores tales como la objetividad, la independencia, la imparcialidad o la transparencia. Es fundamental que los profesionales de la salud, los investigadores y los comités de ética de la investigación garanticen que el avance científico se realice en cumplimiento de los principios éticos y legales, con plenas garantías de los derechos de los participantes en la investigación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
