



La encefalitis de Rasmussen (ER) es una enfermedad rara que afecta fundamentalmente a niños, sin existir diferencias en cuanto al sexo, siendo la edad media de presentación los 7 años. Su etiología continúa siendo desconocida, si bien Rasmussen proponía como desencadenante del cuadro un agente viral, los últimos estudios revelan un probable origen inmunitario mediado por autoanticuerpos y células T citotóxicas1,2.
Presentamos el caso de una niña de 5 años que acudió al servicio de Urgencias por presentar 2 crisis consistentes en movimientos clónicos de miembros superior e inferior derechos de 2 min de duración, sin pérdida de conciencia. No presentaba antecedentes personales de interés y entre los familiares únicamente destacaba la presencia de manchas «café con leche» en la rama paterna.
En la exploración física se evidenciaba una pérdida de fuerza de las extremidades derechas (III/V) y la presencia de 5 manchas «café con leche», siendo el resto de la exploración, por órganos y aparatos, normal.
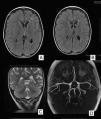
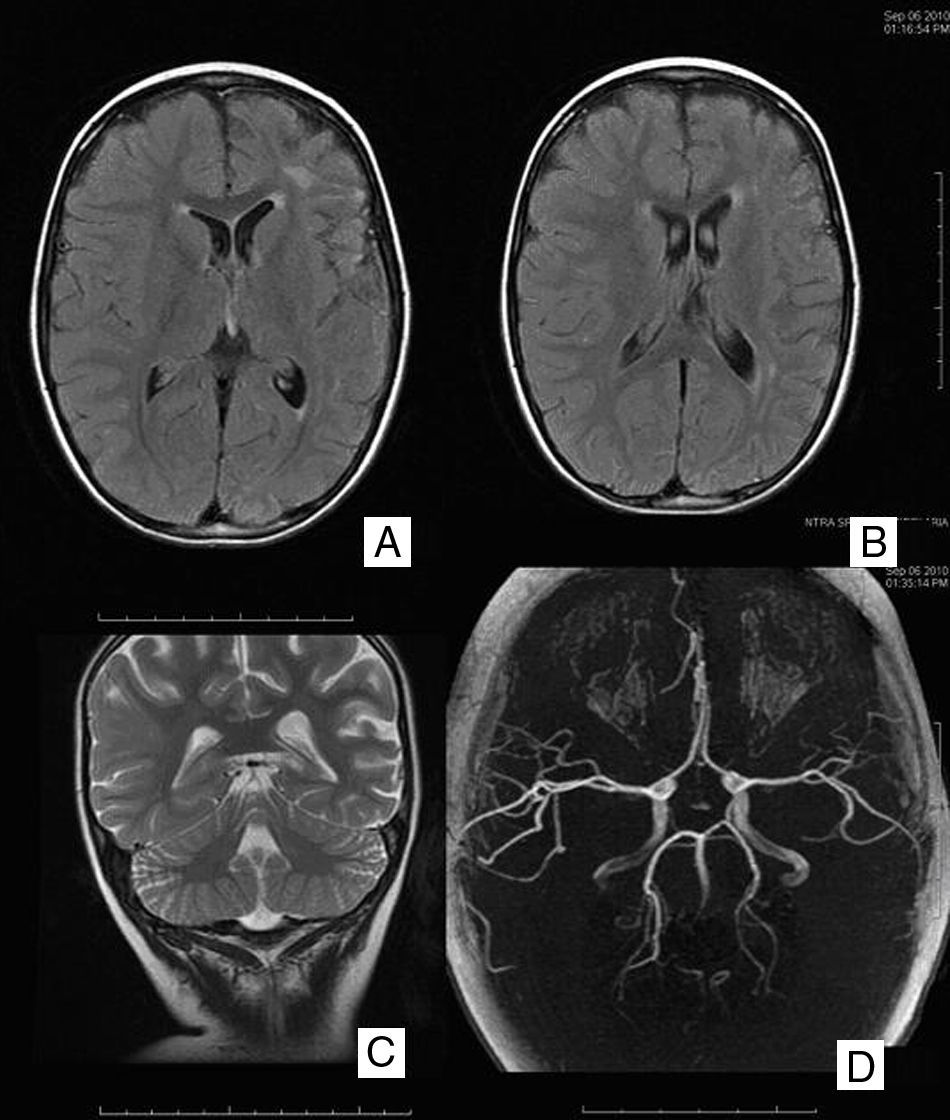
Se realizó un escáner cerebral y un electroencefalograma (EEG) que fueron normales. En la resonancia magnética cerebral (RM) se objetivó un área de alta señal en secuencias T2 afectando a la sustancia blanca paraventricular izquierda, con discreto agrandamiento ventricular y del espacio subaracnoideo hemisférico homolateral (fig. 1A y B). Estos hallazgos sugerían un aspecto residual de la lesión, planteándose como causa un evento isquémico. Fue dada de alta con ácido acetil salicílico y oxcarbacepina con adecuado control de las crisis, persistiendo la hemiparesia derecha.
Reingresó repetidamente en los siguientes 3 meses por crisis de similares características, añadiéndose mioclonías palpebrales y oromentonianas derechas, llegando a precisar ingreso en la Unidad de Cuidados Intensivos por estatus. Recibió tratamiento con varios fármacos antiepiléticos (FAE) (clonacepam, oxcarbacepina, midazolam, etosuximida, valproato, fenitoína, fenobarbital, levetiracetam), sin control de las crisis, siendo diagnosticada de epilepsia parcial continua (EPC).
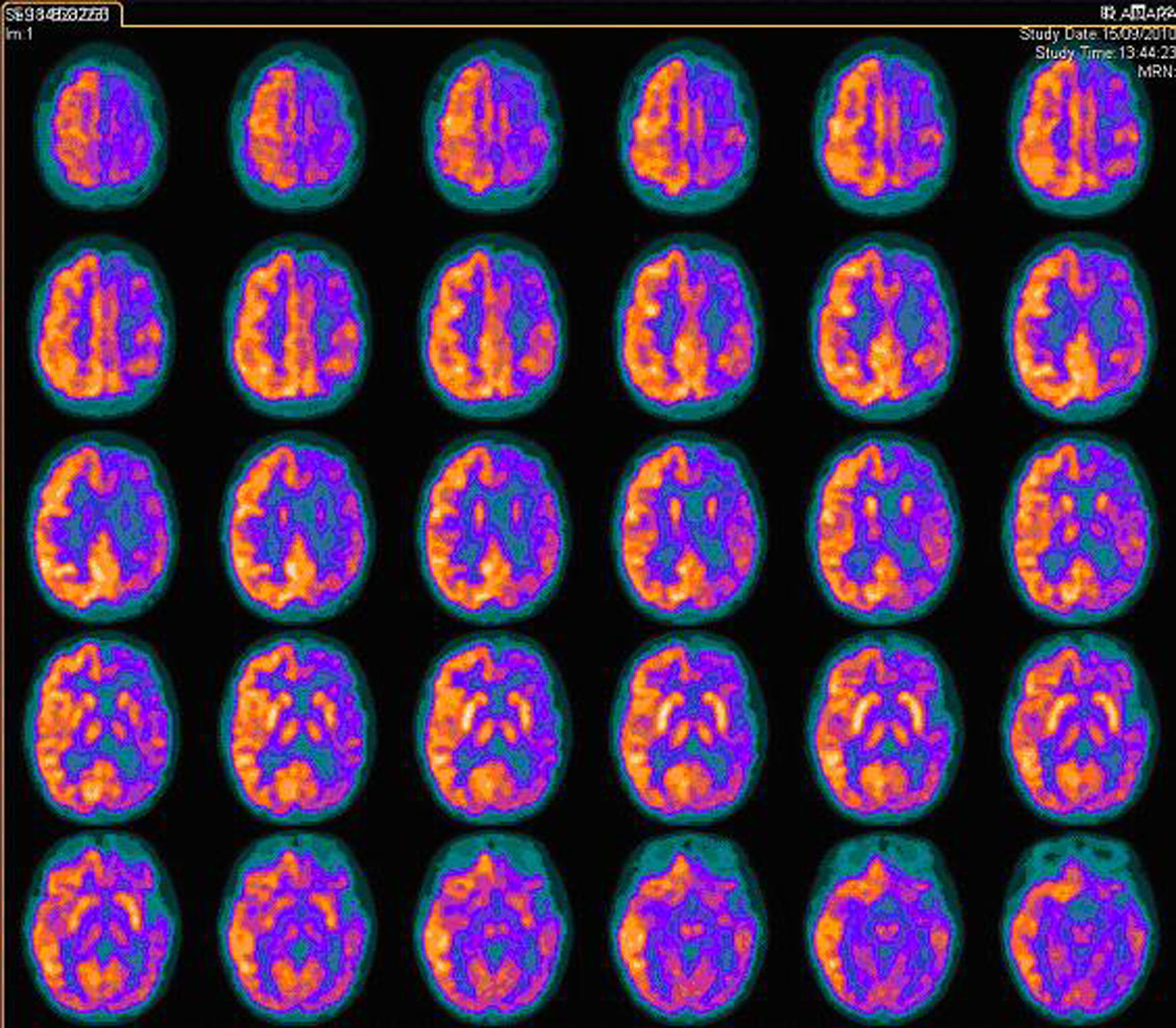
RMc seriadas mostraron un progresivo agrandamiento ventricular y del espacio subaracnoideo hemisférico izquierdo, persistiendo el área de alta señal en la sustancia blanca (fig. 1C). La espectroscopia fue normal. En la secuencia arteriográfica existía una significativa disminución de ramas distales de la arteria cerebral media izquierda respecto de la contralateral, por lo que se realizó una angiografía cerebral convencional que descartó el origen vasculítico (fig. 1D). Finalmente, se practicó una exploración combinada de tomografía por emisión de positrones (PET) y tomografía computarizada (TC), tras la administración por vía intravenosa de 79 MBq de FDG, donde se observó un hipometabolismo moderado-severo del hemisferio cerebral izquierdo con mayor afectación de la región frontal y temporal (fig. 2).
Se realizó una biopsia muscular que mostraba una disminución de la actividad de todos los complejos de la cadena respiratoria mitocondrial. Ante estos hallazgos y la negatividad del resto de pruebas complementarias solicitadas para descartar enfermedad metabólica, autoinmunitaria, infecciosa y protrombótica, se postula el diagnóstico de ER, reorientándose el caso.
Recibió inicialmente pulsos de corticoides a altas dosis (metilprednisolona a 20mg/kg/día) y un ciclo de inmunoglobulinas (0,4g/kg/día) sin mejoría.
En la actualidad, está en tratamiento con prednisona y ciclos mensuales de inmunoglobulinas, además de levetiracetam, clobazam, fenobarbital, coenzima Q10, carnitina y vitaminas, con desaparición de las mioclonías, pero con persistencia de crisis parciales continuas y empeoramiento de la hemiparesia con afectación del lenguaje.
La ER se presenta en individuos con un desarrollo neurológico previo normal, afectando habitualmente a un solo hemisferio, que va sufriendo una atrofia progresiva.
Se manifiesta en 3 estadios: un periodo prodrómico, con escasos signos y síntomas, seguido de la fase aguda con deterioro progresivo de las funciones del hemisferio afectado (hemiparesia, hemianopsia, afasia, deterioro cognitivo) asociado frecuentemente a la aparición de crisis intratables. Hasta en un 50% de los casos se manifiesta como una EPC, también existen casos con escasas crisis o incluso sin ellas. Tras un periodo de unos 8 meses aparece la fase de estado con estabilización del deterioro neurológico y disminución del número de crisis3.
El diagnóstico se basa en la clínica junto con los hallazgos de estudios neurofisiológicos y de imagen, quedando establecidos los criterios diagnósticos en el Consenso Europeo del 20052. Se debe realizar el diagnóstico diferencial con otras causas de hemiatrofia cerebral, como pueden ser el síndrome de Sturge-Weber, el infarto hemisférico (Dyke-Davidoff-Masson) y el síndrome de MELAS.
Nuestra paciente presentó EPC y hemiparesia, lo que facilitó el diagnóstico junto con los estudios de imagen seriados que demostraban la progresiva atrofia hemisférica. Se descartó el origen isquémico y, por estudio genético del ADN mitocondrial, la mutación A-G en el nucleótido 3243 (tRNA Leu (UUR)), presente en el 80% de los casos de MELAS. Tampoco se encontraron mutaciones patológicas en el gen tRNA (Leu(UUR)) ni depleción.
Desde el punto de vista terapéutico, hay que tener en cuenta 2 aspectos fundamentales: el tratamiento de las crisis, con muy pobre respuesta en general a los FAE, siendo la hemisferectomía funcional la alternativa más satisfactoria y el intento de frenar el deterioro neurológico, para lo que se emplean inmunoglobulinas por vía intravenosa, corticoides a dosis altas e incluso plasmaféresis4,5. Teniendo en cuenta que la hemisferectomía funcional previene también del deterioro cognitivo, secundariamente al controlar las crisis6, las indicaciones se deben individualizar en cada caso, tomando decisiones en consenso con los pacientes y/o familiares y sopesando la severidad de los déficits esperados tras la hemisferectomía con la severidad de la epilepsia y los potenciales efectos secundarios del tratamiento farmacológico7. La cirugía suele posponerse hasta que la hemiparesia está instaurada.
En nuestro caso, ha sido derivada al hospital de referencia para estudio de cirugía de la epilepsia debido a la escasa respuesta a FAE y al progresivo deterioro neurológico pese al tratamiento.
